Joint single-cell mitochondrial DNA genotyping and DNA accessibility analysis
Caleb Lareau and Tim Stuart
Compiled: April 28, 2021
Source:vignettes/mito.Rmd
mito.RmdHere, we take a look at two different datasets containing both DNA accessibility measurements and mitochondrial mutation data in the same cells. One was sampled from a patient with a colorectal cancer (CRC) tumor, and the other is from a polyclonal TF1 cell line. This data was produced by Lareau and Ludwig et al. (2020), and you can read the original paper here: https://doi.org/10.1038/s41587-020-0645-6.
Processed data files, including mitochondrial variant data for the CRC and TF1 dataset is available on Zenodo here: https://zenodo.org/record/3977808
Raw sequencing data and DNA accessibility processed files for the these datasets are available on NCBI GEO here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142745
Colorectal cancer dataset
To demonstrate combined analyses of mitochondrial DNA variants and accessible chromatin, we’ll walk through a vignette analyzing cells from a primary colorectal adenocarcinoma. The sample contains a mixture of malignant epithelial cells and tumor infiltrating immune cells.
Loading the DNA accessibility data
First we load the scATAC-seq data and create a Seurat object following the standard workflow for scATAC-seq data.
# load counts and metadata from cellranger-atac
counts <- Read10X_h5(filename = "../vignette_data/mito/CRC_v12-mtMask_mgatk.filtered_peak_bc_matrix.h5")
metadata <- read.csv(
file = "../vignette_data/mito/CRC_v12-mtMask_mgatk.singlecell.csv",
header = TRUE,
row.names = 1
)
# load gene annotations from Ensembl
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)
# change to UCSC style since the data was mapped to hg19
seqlevelsStyle(annotations) <- 'UCSC'
genome(annotations) <- "hg19"
# create object
crc_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
annotation = annotations,
min.cells = 10,
genome = "hg19",
fragments = '../vignette_data/mito/CRC_v12-mtMask_mgatk.fragments.tsv.gz'
)
crc <- CreateSeuratObject(
counts = crc_assay,
assay = 'peaks',
meta.data = metadata
)
crc[["peaks"]]## ChromatinAssay data with 81787 features for 3535 cells
## Variable features: 0
## Genome: hg19
## Annotation present: TRUE
## Motifs present: FALSE
## Fragment files: 1Quality control
We can compute the standard quality control metrics for scATAC-seq and filter out low-quality cells based on these metrics.
# Augment QC metrics that were computed by cellranger-atac
crc$pct_reads_in_peaks <- crc$peak_region_fragments / crc$passed_filters * 100
crc$pct_reads_in_DNase <- crc$DNase_sensitive_region_fragments / crc$passed_filters * 100
crc$blacklist_ratio <- crc$blacklist_region_fragments / crc$peak_region_fragments
# compute TSS enrichment score and nucleosome banding pattern
crc <- TSSEnrichment(crc)
crc <- NucleosomeSignal(crc)
# visualize QC metrics for each cell
VlnPlot(crc, c("TSS.enrichment", "nCount_peaks", "nucleosome_signal", "pct_reads_in_peaks", "pct_reads_in_DNase", "blacklist_ratio"), pt.size = 0, ncol = 3)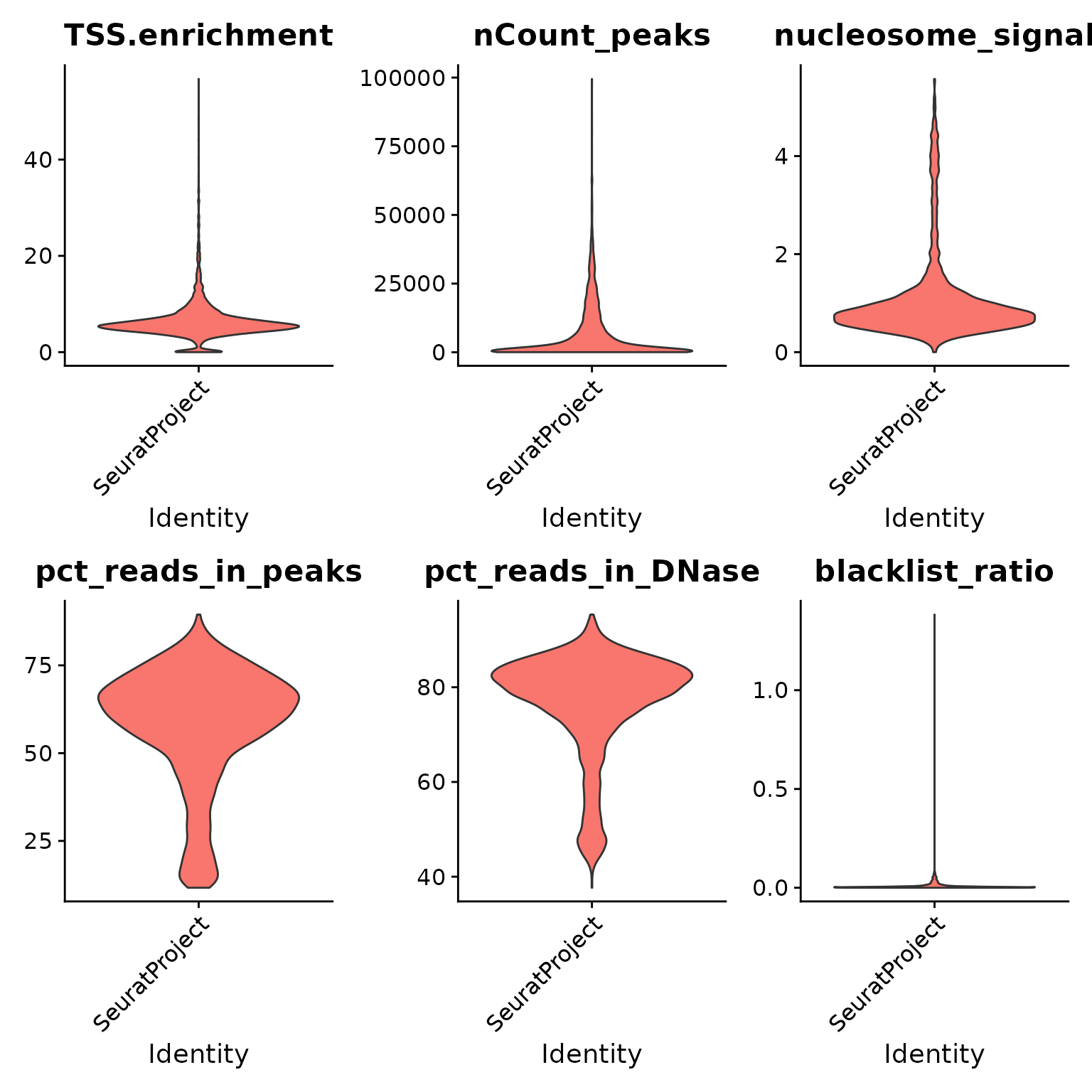
# remove low-quality cells
crc <- subset(
x = crc,
subset = nCount_peaks > 1000 &
nCount_peaks < 50000 &
pct_reads_in_DNase > 40 &
blacklist_ratio < 0.05 &
TSS.enrichment > 3 &
nucleosome_signal < 4
)
crc## An object of class Seurat
## 81787 features across 1861 samples within 1 assay
## Active assay: peaks (81787 features, 0 variable features)Loading the mitochondrial variant data
Next we can load the mitochondrial DNA variant data for these cells that was quantified using mgatk. The ReadMGATK() function in Signac allows the output from mgatk to be read directly into R in a convenient format for downstream analysis with Signac. Here, we load the data and add it to the Seurat object as a new assay.
# load mgatk output
mito.data <- ReadMGATK(dir = "../vignette_data/mito/crc/")
# create an assay
mito <- CreateAssayObject(counts = mito.data$counts)
# Subset to cell present in the scATAC-seq assat
mito <- subset(mito, cells = colnames(crc))
# add assay and metadata to the seurat object
crc[["mito"]] <- mito
crc <- AddMetaData(crc, metadata = mito.data$depth, col.name = "mtDNA_depth")We can look at the mitochondrial sequencing depth for each cell, and further subset the cells based on mitochondrial sequencing depth.
VlnPlot(crc, "mtDNA_depth", pt.size = 0.1) + scale_y_log10()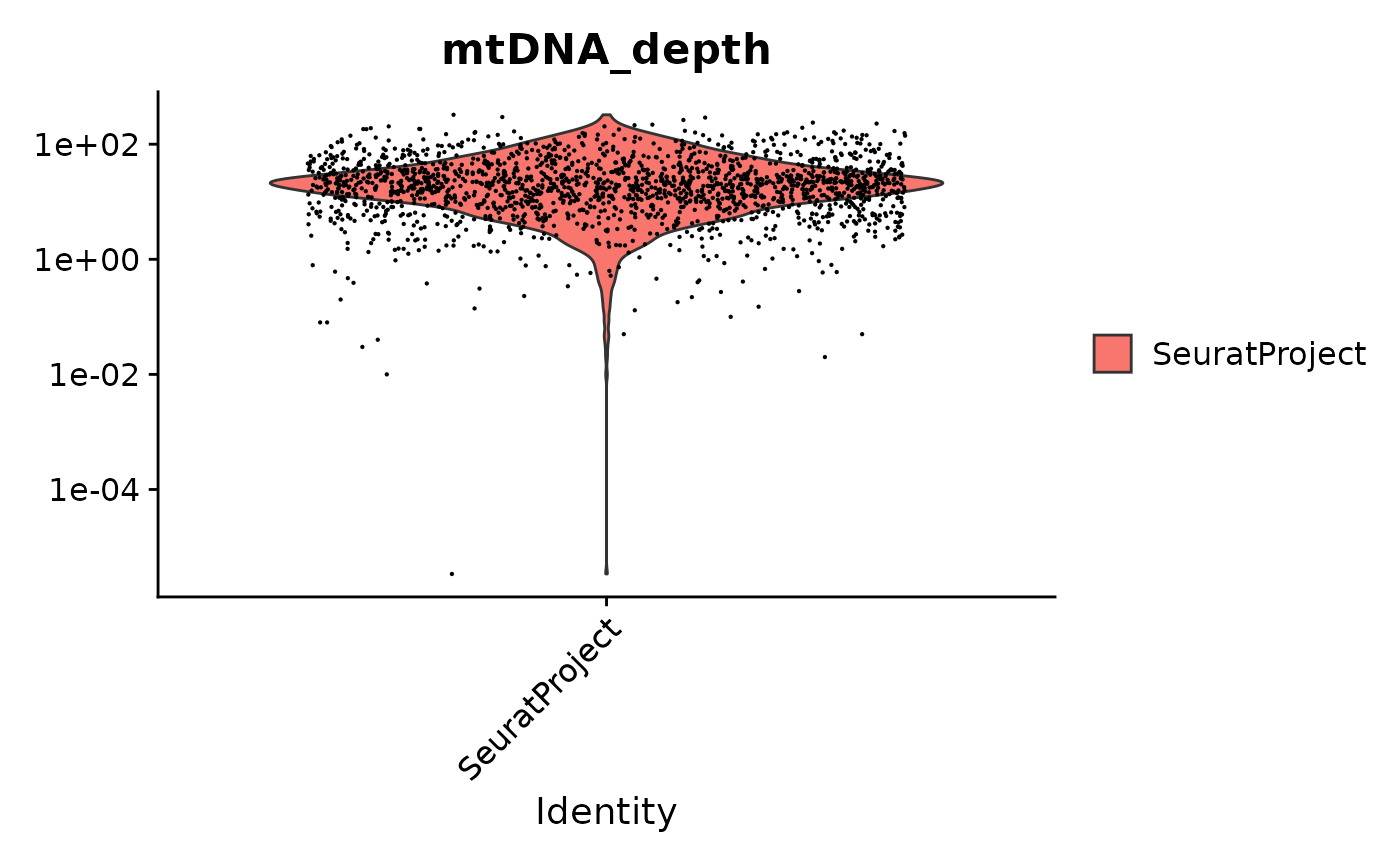
# filter cells based on mitochondrial depth
crc <- subset(crc, mtDNA_depth >= 10)
crc## An object of class Seurat
## 214339 features across 1372 samples within 2 assays
## Active assay: peaks (81787 features, 0 variable features)
## 1 other assay present: mitoDimension reduction and clustering
Next we can run a standard dimension reduction and clustering workflow using the scATAC-seq data to identify cell clusters.
crc <- RunTFIDF(crc)
crc <- FindTopFeatures(crc, min.cutoff = 10)
crc <- RunSVD(crc)
crc <- RunUMAP(crc, reduction = "lsi", dims = 2:50)
crc <- FindNeighbors(crc, reduction = "lsi", dims = 2:50)
crc <- FindClusters(crc, resolution = 0.5, algorithm = 3)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 1372
## Number of edges: 62089
##
## Running smart local moving algorithm...
## Maximum modularity in 10 random starts: 0.8108
## Number of communities: 6
## Elapsed time: 0 seconds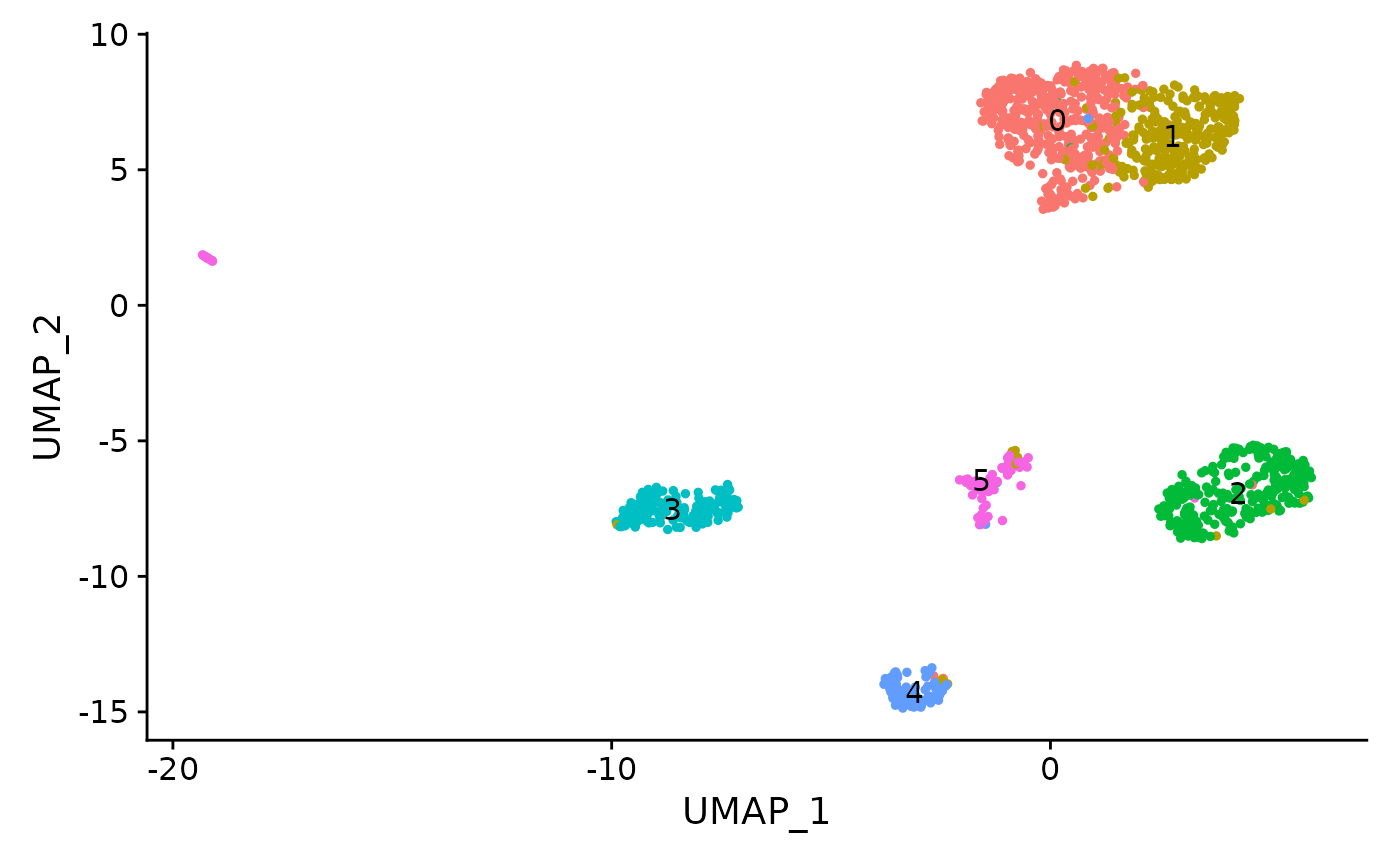
Generate gene scores
To help interpret these clusters of cells, and assign a cell type label, we’ll estimate gene activities by summing the DNA accessibility in the gene body and promoter region.
# compute gene accessibility
gene.activities <- GeneActivity(crc)
# add to the Seurat object as a new assay
crc[['RNA']] <- CreateAssayObject(counts = gene.activities)
crc <- NormalizeData(
object = crc,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(crc$nCount_RNA)
)Visualize interesting gene activity scores
We note the following markers for different cell types in the CRC dataset:
- EPCAM is a marker for epithelial cells
- TREM1 is a meyloid marker
- PTPRC = CD45 is a pan-immune cell marker
- IL1RL1 is a basophil marker
- GATA3 is a Tcell maker
DefaultAssay(crc) <- 'RNA'
FeaturePlot(
object = crc,
features = c('TREM1', 'EPCAM', "PTPRC", "IL1RL1","GATA3", "KIT"),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 2
)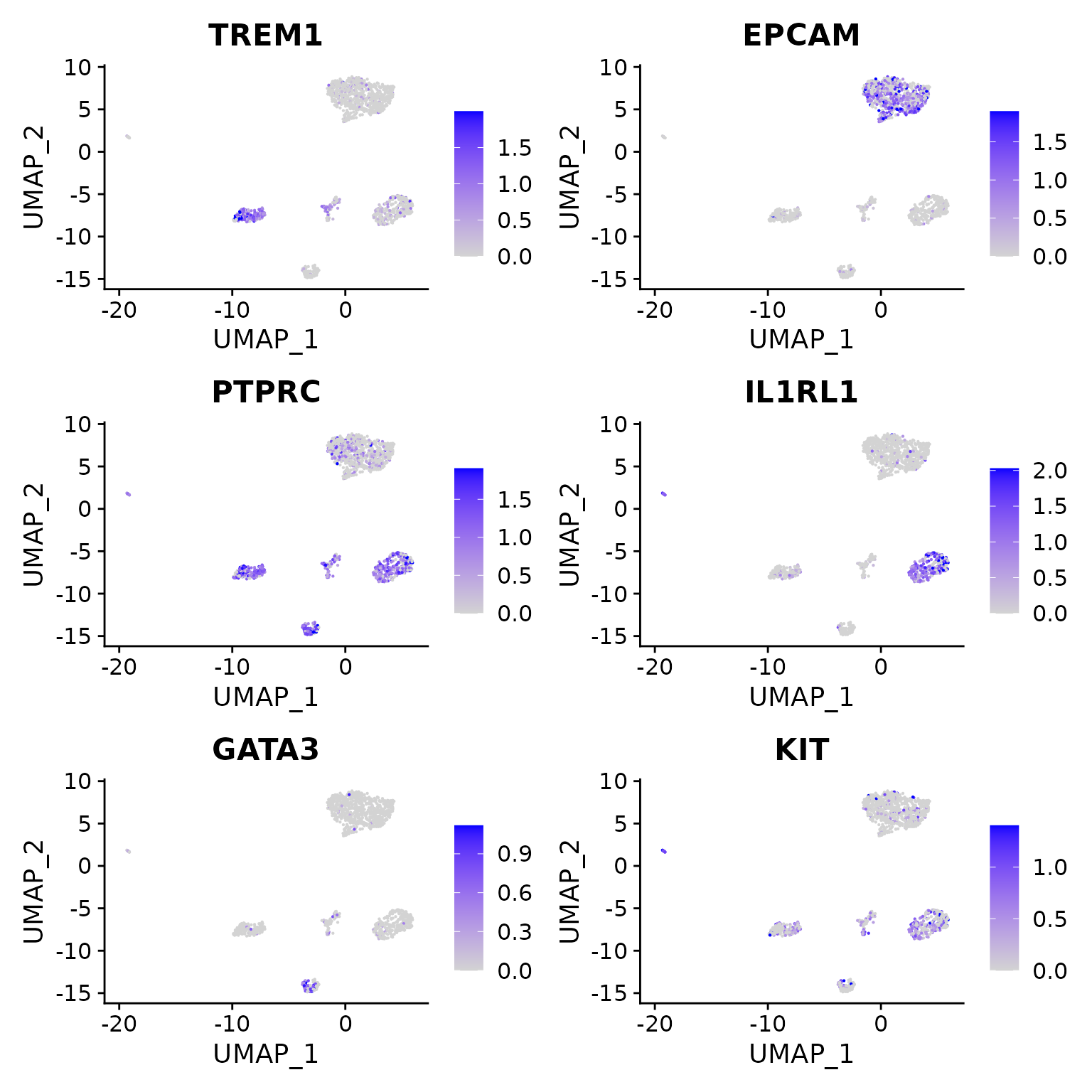
Using these gene score values, we can assign cluster identities:
crc <- RenameIdents(
object = crc,
'0' = 'Epithelial',
'1' = 'Epithelial',
'2' = 'Basophil',
'3' = 'Myeloid_1',
'4' = 'Myeloid_2',
'5' = 'Tcell'
)One of the myeloid clusters has a lower percentage of fragments in peaks, as well as a lower overall mitochondrial sequencing depth and a different nucleosome banding pattern.
p1 <- FeatureScatter(crc, "mtDNA_depth", "pct_reads_in_peaks") + ggtitle("") + scale_x_log10()
p2 <- FeatureScatter(crc, "mtDNA_depth", "nucleosome_signal") + ggtitle("") + scale_x_log10()
p1 + p2 + plot_layout(guides = 'collect')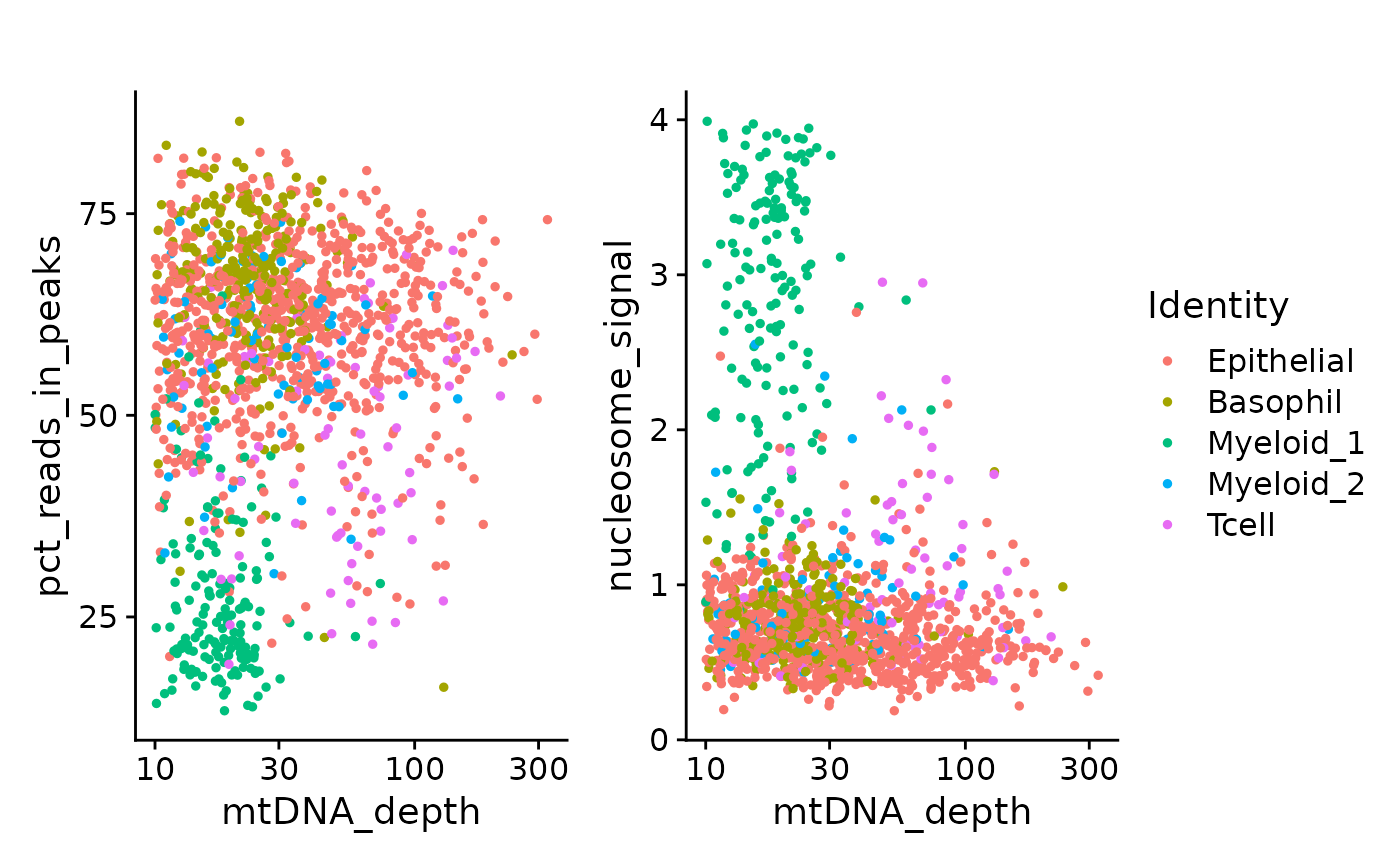
We can see that most of the low FRIP cells were the myeloid 1 cluster. This is most likely an intra-tumor granulocyte that has relatively poor accessible chromatin enrichment. Similarly, the unusual nuclear chromatin packaging of this cell type yields slightly reduced mtDNA coverage compared to the myeloid 2 cluster.
Find informative mtDNA variants
Next, we can identify sites in the mitochondrial genome that vary across cells, and cluster the cells into clonotypes based on the frequency of these variants in the cells. Signac utilizes the principles established in the original mtscATAC-seq work of identifying high-quality variants.
variable.sites <- IdentifyVariants(crc, assay = "mito", refallele = mito.data$refallele)
VariantPlot(variants = variable.sites)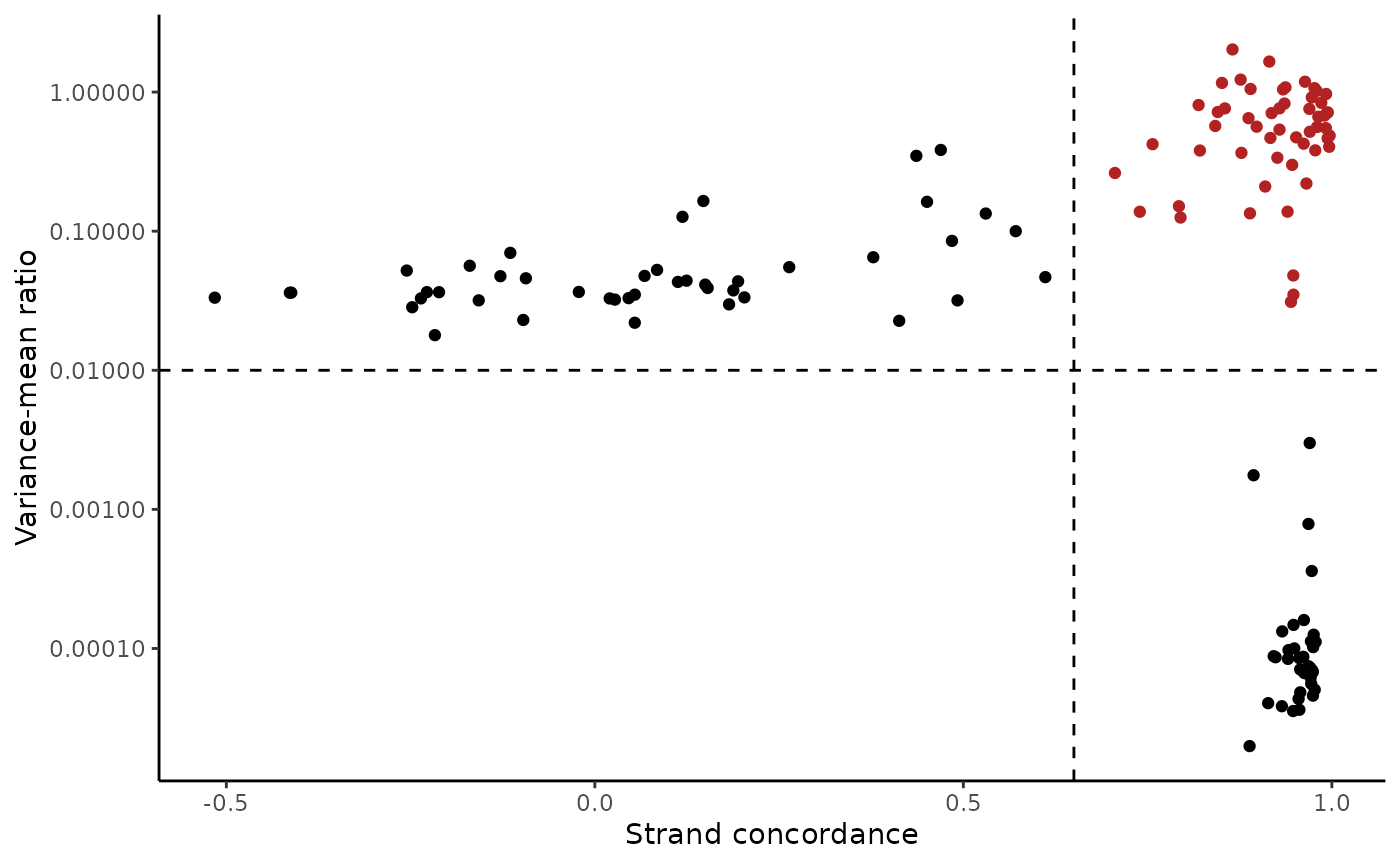
The plot above clearly shows a group of variants with a higher VMR and strand concordance. In principle, a high strand concordance reduces the likelihood of the allele frequency being driven by sequencing error (which predominately occurs on one but not the other strand. This is due to the preceding nucleotide content and a common error in mtDNA genotyping). On the other hand, variants that have a high VMR are more likely to be clonal variants as the alternate alleles tend to aggregate in certain cells rather than be equivalently dispersed about all cells, which would be indicative of some other artifact.
We note that variants that have a very low VMR and and very high strand concordance are homoplasmic variants for this sample. While these may be interesting in some settings (e.g. donor demultiplexing), for inferring subclones, these are not particularly useful.
Based on these thresholds, we can filter out a set of informative mitochondrial variants that differ across the cells.
# Establish a filtered data frame of variants based on this processing
high.conf <- subset(
variable.sites, subset = n_cells_conf_detected >= 5 &
strand_correlation >= 0.65 &
vmr > 0.01
)
high.conf[,c(1,2,5)]## position nucleotide mean
## 1227G>A 1227 G>A 0.0083414
## 3244G>A 3244 G>A 0.0012223
## 6081G>A 6081 G>A 0.0027369
## 9804G>A 9804 G>A 0.0032605
## 12889G>A 12889 G>A 0.0226331
## 9728C>T 9728 C>T 0.0133554
## 16147C>T 16147 C>T 0.6405975
## 824T>C 824 T>C 0.0054360
## 2285T>C 2285 T>C 0.0055185
## 9840T>C 9840 T>C 0.0021229
## 16093T>C 16093 T>C 0.0079146A few things stand out. First, 10 out of the 12 variants occur at less than 1% allele frequency in the population. However, 16147C>T is present at about 62%. We’ll see that this is a clonal variant marking the epithelial cells. Additionally, all of the called variants are transitions (A - G or C - T) rather than transversion mutations (A - T or C - G). This fits what we know about how these mutations arise in the mitochondrial genome.
Depending on your analytical question, these thresholds can be adjusted to identify variants that are more prevalent in other cells.
Compute the variant allele frequency for each cell
We currently have information for each strand stored in the mito assay to allow strand concordance to be assessed. Now that we have our set of high-confidence informative variants, we can create a new assay containing strand-collapsed allele frequency counts for each cell for these variants using the AlleleFreq() function.
crc <- AlleleFreq(
object = crc,
variants = high.conf$variant,
assay = "mito"
)
crc[["alleles"]]## Assay data with 11 features for 1372 cells
## First 10 features:
## 1227G>A, 3244G>A, 6081G>A, 9804G>A, 12889G>A, 9728C>T, 16147C>T,
## 824T>C, 2285T>C, 9840T>CVisualize the variants
Now that the allele frequencies are stored as an additional assay, we can use the standard functions in Seurat to visualize how these allele frequencies are distributed across the cells. Here we visualize a subset of the variants using FeaturePlot() and DoHeatmap().
DefaultAssay(crc) <- "alleles"
alleles.view <- c("12889G>A", "16147C>T", "9728C>T", "9804G>A")
FeaturePlot(
object = crc,
features = alleles.view,
order = TRUE,
cols = c("grey", "darkred"),
ncol = 4
) & NoLegend()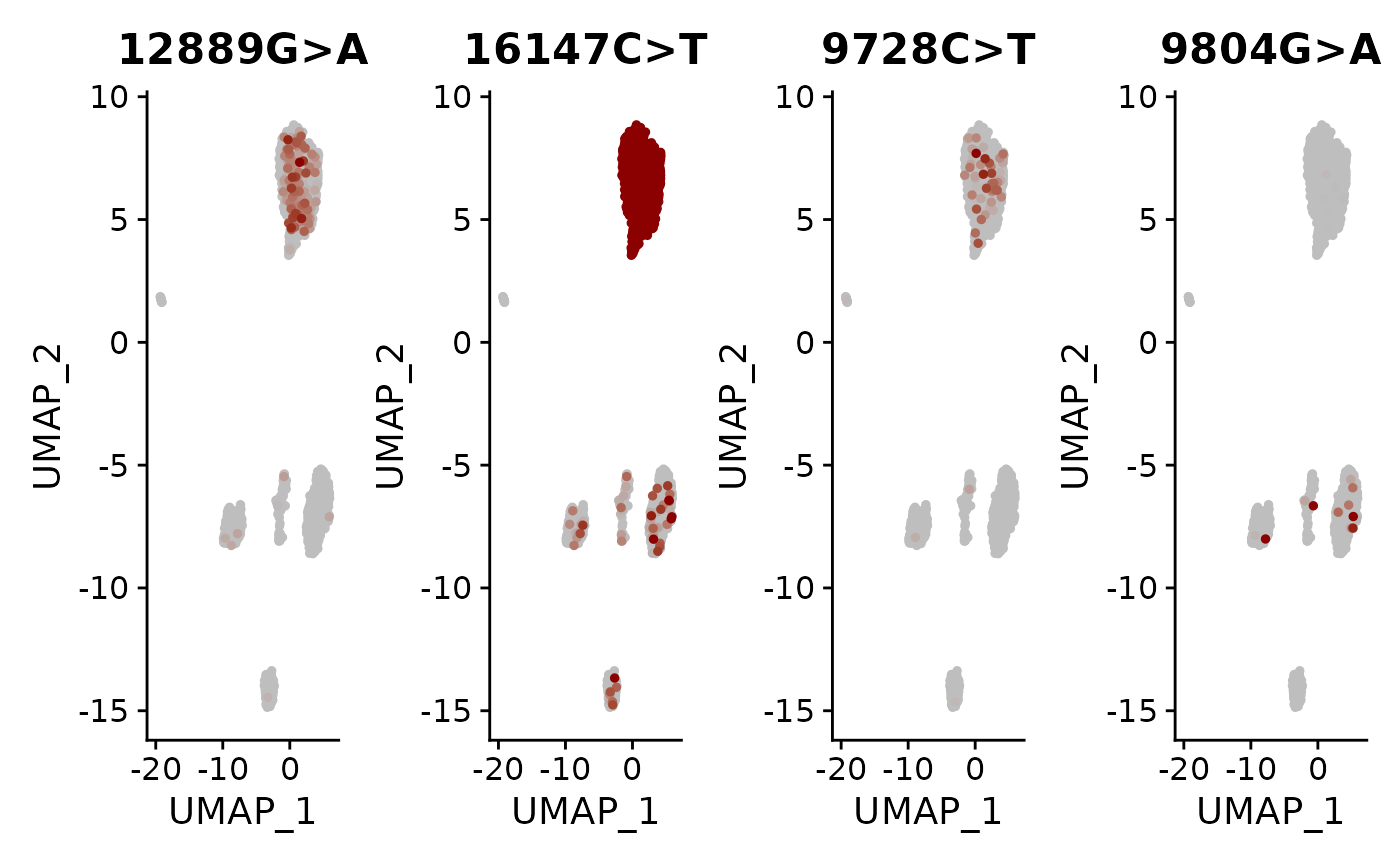
DoHeatmap(crc, features = rownames(crc), slot = "data", disp.max = 1) +
scale_fill_viridis_c()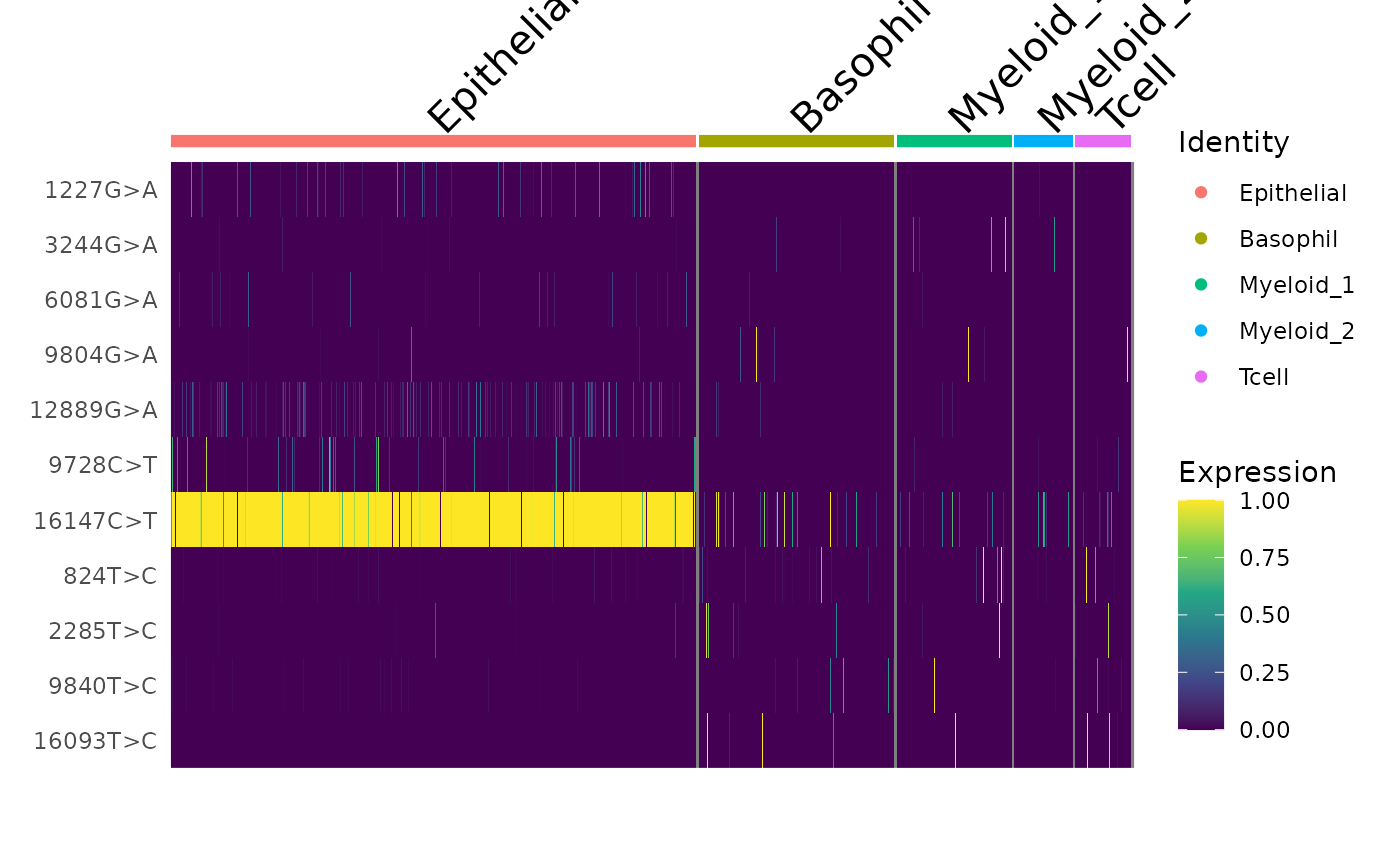
Here, we can see a few interesting patterns for the selected variants. 16147C>T is present in essentially all epithelial cells and almost exclusively in epithelial cells (the edge cases where this isn’t true are also cases where the UMAP and clustering don’t full agree). It is at 100% allele frequency– strongly suggestive of whatever cell of origin of this tumor had the mutation at 100% and then expanded. We then see at least 3 variants 1227G>A, 12889G>A, and 9728C>T that are mostly present specifically in the epithelial cells that define subclones. Other variants including 3244G>A, 9804G>A, and 824T>C are found specifically in immune cell populations, suggesting that these arose from a common hematopoetic progenitor cell (probably in the bone marrow).
TF1 cell line dataset
Next we’ll demonstrate a similar workflow to identify cell clones in a different dataset, this time generated from a TF1 cell line. This dataset contains more clones present at a higher proportion, based on the experimental design.
We’ll demonstrate how to identify groups of related cells (clones) by clustering the allele frequency data and how to relate these clonal groups to accessibility differences utilizing the multimodal capabilities of Signac.
Data loading
# read the mitochondrial data
tf1.data <- ReadMGATK(dir = "../vignette_data/mito/tf1/")## Reading allele counts## Reading metadata## Building matrices
# create a Seurat object
tf1 <- CreateSeuratObject(
counts = tf1.data$counts,
meta.data = tf1.data$depth,
assay = "mito"
)
# load the peak set
peaks <- read.table(
file = "../vignette_data/mito/TF1.filtered.narrowPeak.gz",
sep = "\t",
col.names = c("chrom", "start", "end", "peak", "width", "strand", "x", "y", "z", "w")
)
peaks <- makeGRangesFromDataFrame(peaks)
# create fragment object
frags <- CreateFragmentObject(
path = "../vignette_data/mito/TF1.filtered.fragments.tsv.gz",
cells = colnames(tf1)
)## Computing hash
# quantify the DNA accessibility data
counts <- FeatureMatrix(
fragments = frags,
features = peaks,
cells = colnames(tf1)
)## Extracting reads overlapping genomic regions
# create assay with accessibility data and add it to the Seurat object
tf1[["peaks"]] <- CreateChromatinAssay(
counts = counts,
fragments = frags
)Quality control
# add annotations
Annotation(tf1[["peaks"]]) <- annotations
DefaultAssay(tf1) <- "peaks"
tf1 <- NucleosomeSignal(tf1)
tf1 <- TSSEnrichment(tf1)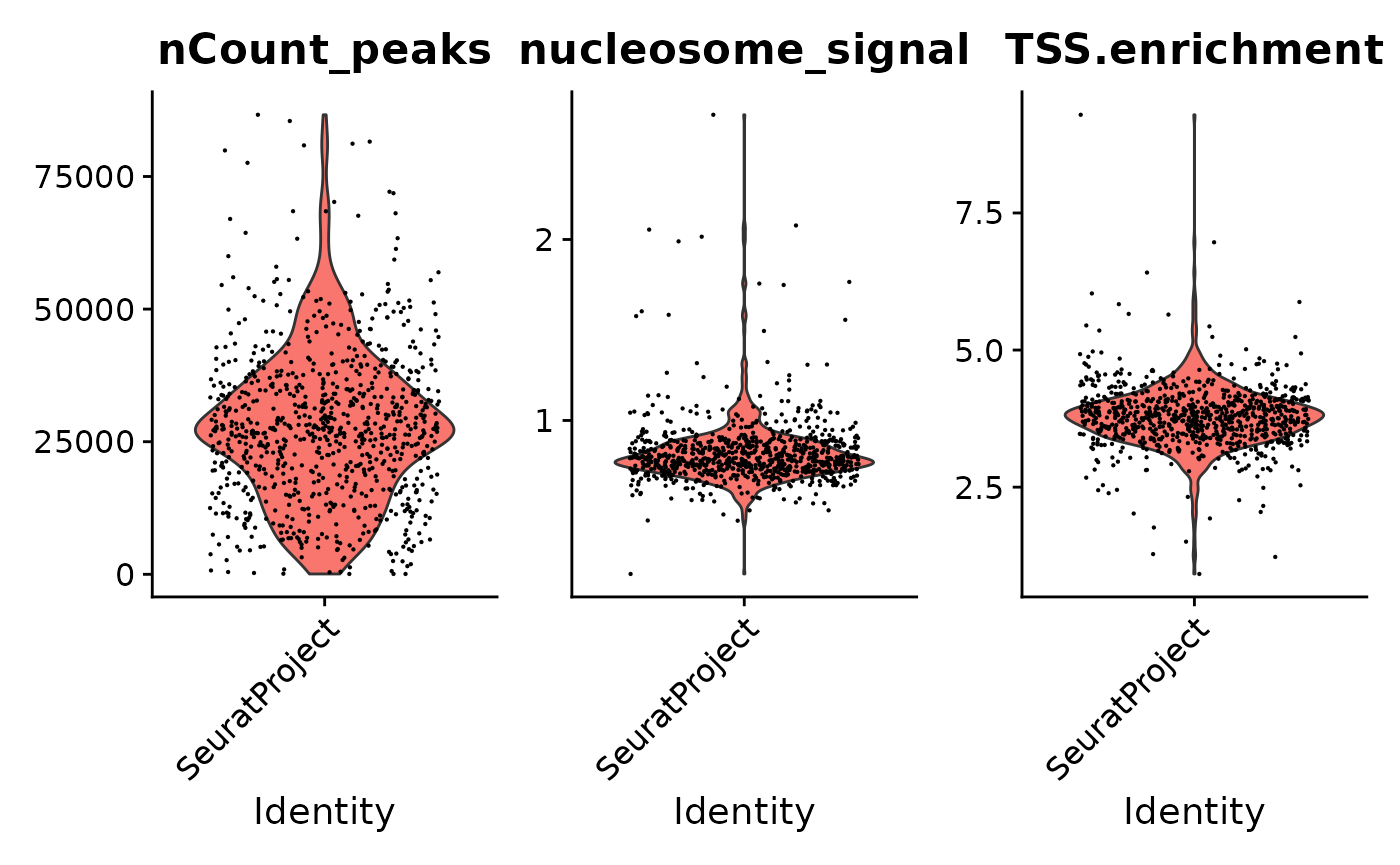
tf1 <- subset(
x = tf1,
subset = nCount_peaks > 500 &
nucleosome_signal < 2 &
TSS.enrichment > 2.5
)
tf1## An object of class Seurat
## 255300 features across 832 samples within 2 assays
## Active assay: peaks (122748 features, 0 variable features)
## 1 other assay present: mitoIdentifying variants
DefaultAssay(tf1) <- "mito"
variants <- IdentifyVariants(tf1, refallele = tf1.data$refallele)## Computing total coverage per base## Processing A## Processing T## Processing C## Processing G
VariantPlot(variants)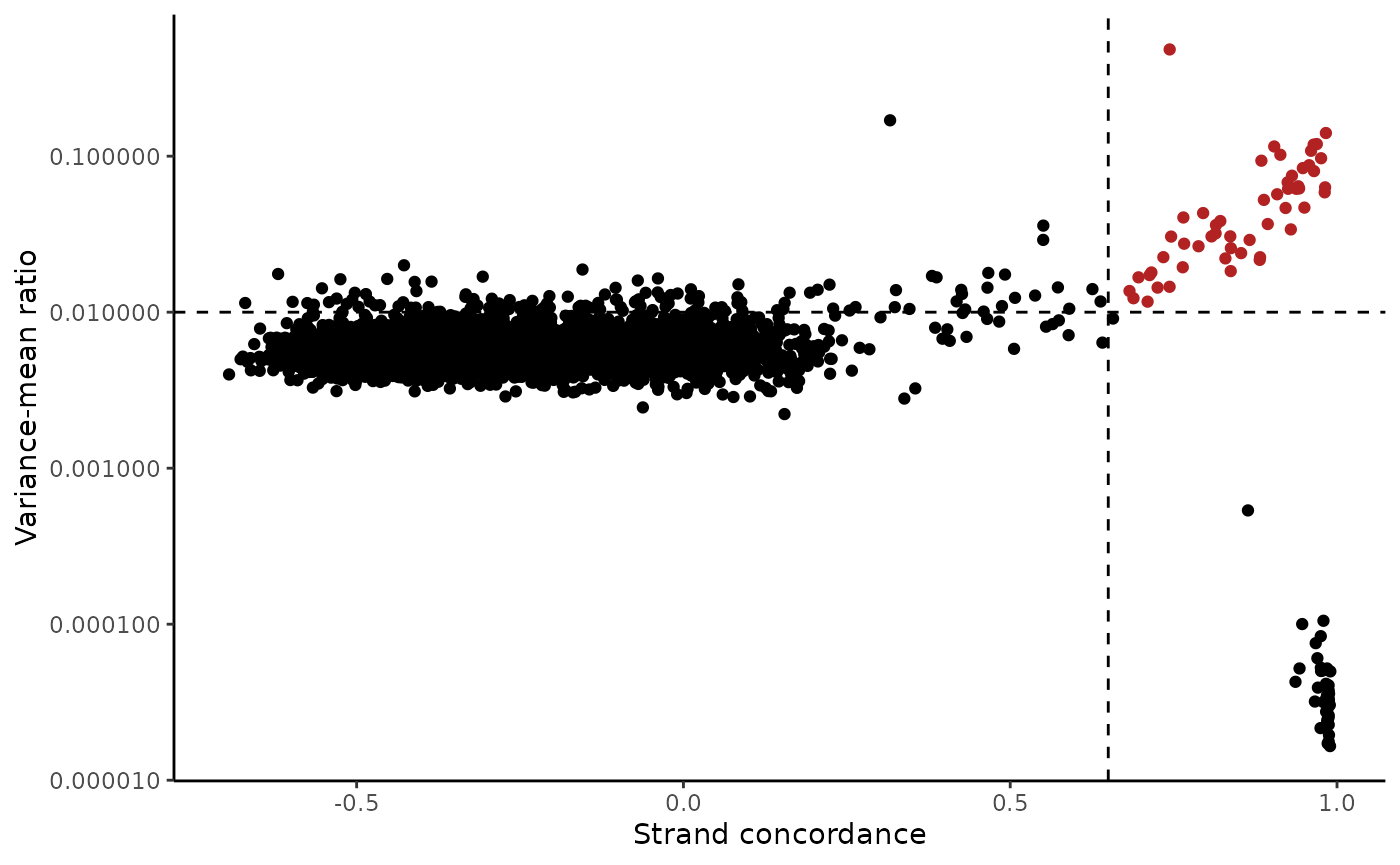
high.conf <- subset(
variants, subset = n_cells_conf_detected >= 5 &
strand_correlation >= 0.65 &
vmr > 0.01
)
tf1 <- AlleleFreq(tf1, variants = high.conf$variant, assay = "mito")
tf1[["alleles"]]## Assay data with 51 features for 832 cells
## First 10 features:
## 627G>A, 709G>A, 1045G>A, 1793G>A, 1888G>A, 1906G>A, 2002G>A, 2040G>A,
## 2573G>A, 2643G>AIdentifying clones
Now that we’ve identified a set of variable alleles, we can cluster the cells based on the frequency of each of these alleles using the FindClonotypes() function. This uses the Louvain community detection algorithm implemented in Seurat.
DefaultAssay(tf1) <- "alleles"
tf1 <- FindClonotypes(tf1)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 832
## Number of edges: 15680
##
## Running smart local moving algorithm...
## Maximum modularity in 10 random starts: 0.8398
## Number of communities: 12
## Elapsed time: 0 seconds##
## 10 11 9 4 7 8 2 3 1 5 0 6
## 17 11 23 107 32 30 116 107 123 80 153 33Here we see that the clonal clustering has identified 12 different clones in the TF1 dataset. We can further visualize the frequency of alleles in these clones using DoHeatmap(). The FindClonotypes() function also performs hierarchical clustering on both the clonotypes and the alleles, and sets the factor levels for the clonotypes based on the hierarchical clustering order, and the order of variable features based on the hierarchical feature clustering. This allows us to get a decent ordering of both features and clones automatically:
DoHeatmap(tf1, features = VariableFeatures(tf1), slot = "data", disp.max = 0.1) +
scale_fill_viridis_c()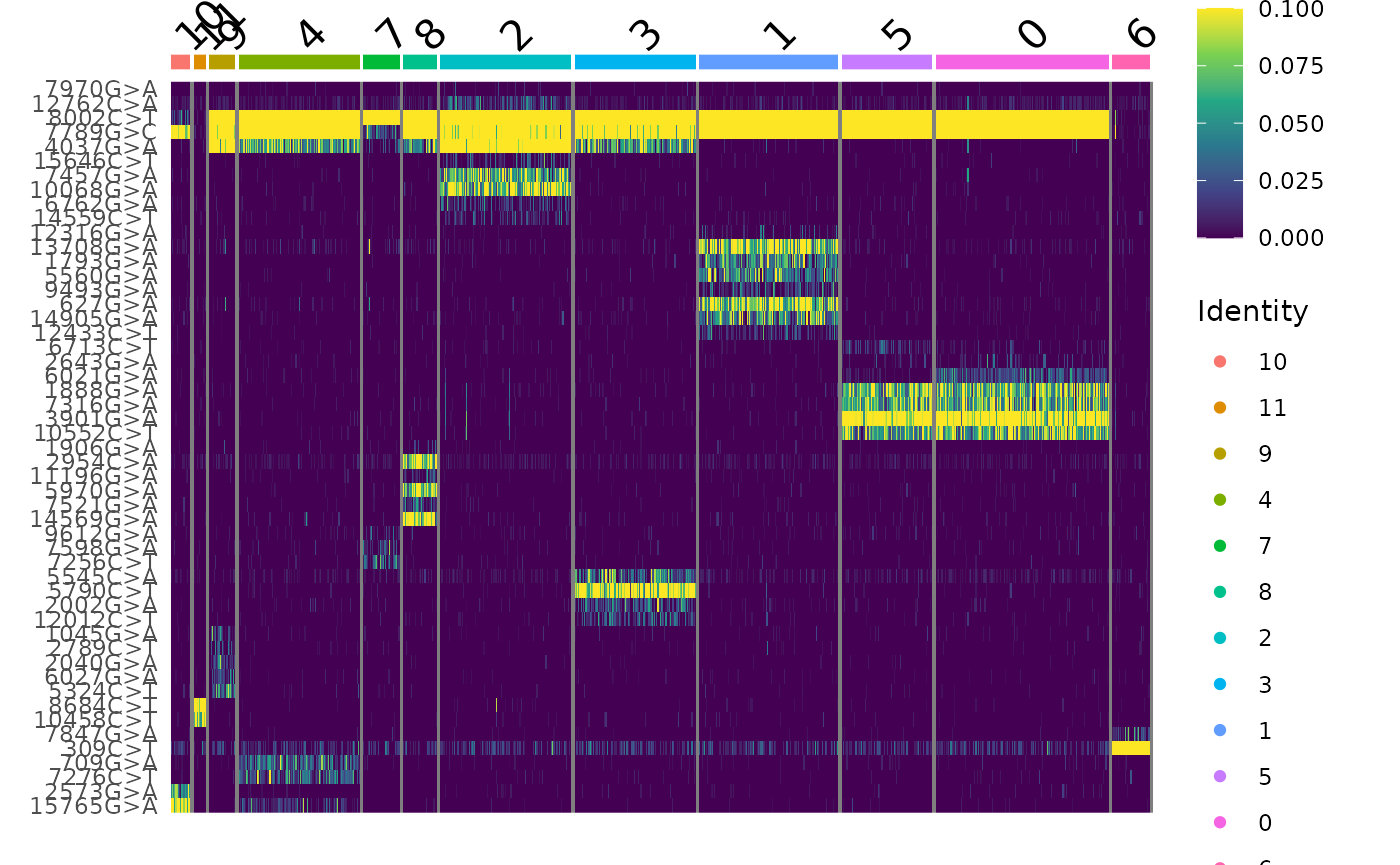
Find differentially accessible peaks between clones
Next we can use the clonal information derived from the mitochondrial assay to find peaks that are differentially accessible between clones.
DefaultAssay(tf1) <- "peaks"
# find peaks specific to one clone
markers.fast <- FoldChange(tf1, ident.1 = 2)
head(markers.fast)## avg_log2FC pct.1 pct.2
## chr1-115658-115894 -0.07337271 0.034 0.063
## chr1-564543-564795 -7.44683695 0.776 0.870
## chr1-565293-565510 -4.59454090 0.603 0.585
## chr1-565875-566090 -14.48377274 0.672 0.578
## chr1-567821-568039 -14.37220215 0.043 0.311
## chr1-569356-569603 -4.88624299 0.802 0.777We can the DNA accessibility in these regions for each clone using the CoveragePlot() function. As you can see, the peaks identified are highly specific to one clone.
CoveragePlot(
object = tf1,
region = rownames(markers.fast)[1],
extend.upstream = 2000,
extend.downstream = 2000
)## Warning: Removed 3 rows containing missing values (position_stack).## Warning: Removed 13 rows containing missing values (geom_segment).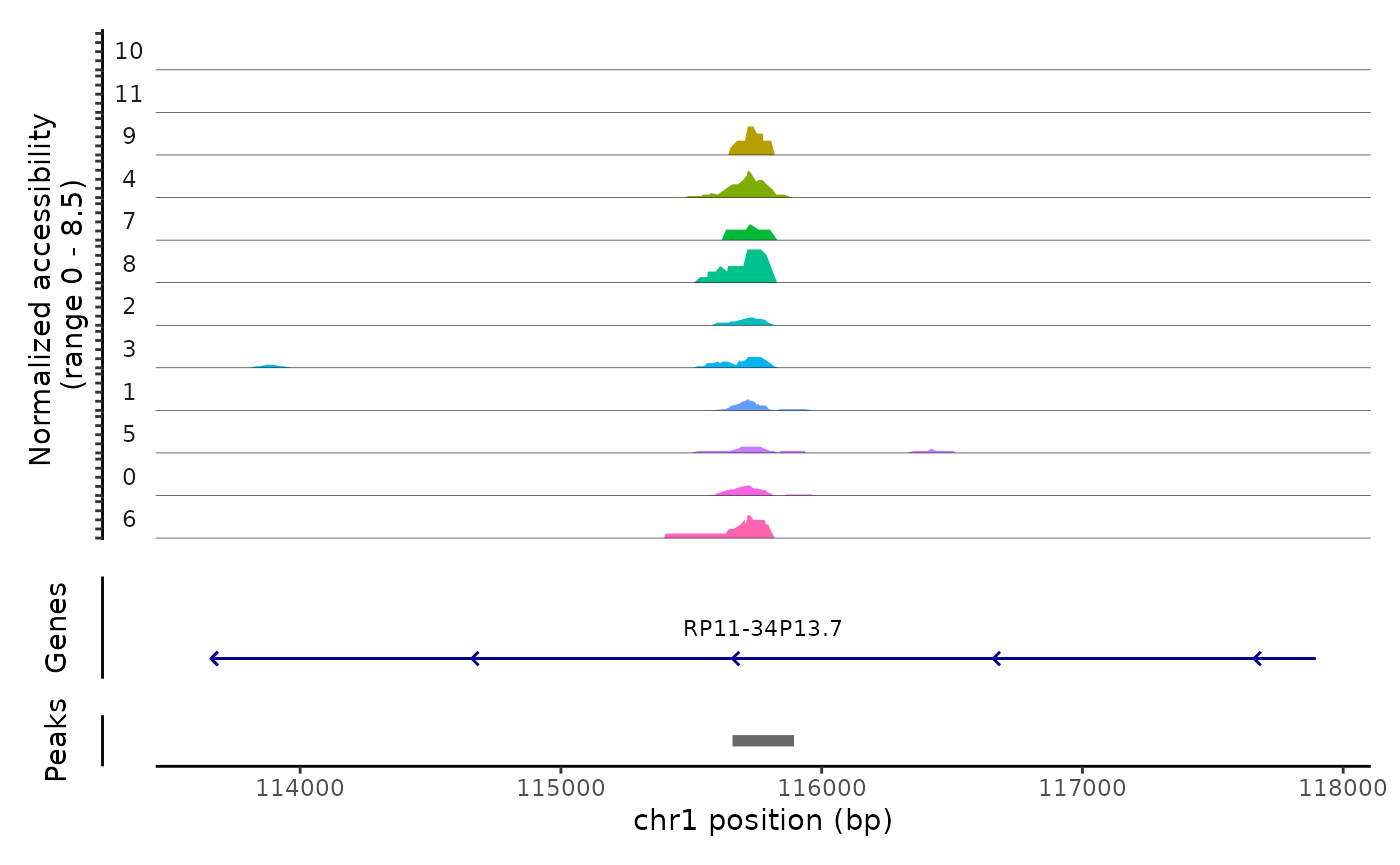
Session Info
## R version 4.0.1 (2020-06-06)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] EnsDb.Hsapiens.v75_2.99.0 ensembldb_2.14.0
## [3] AnnotationFilter_1.14.0 GenomicFeatures_1.42.3
## [5] AnnotationDbi_1.52.0 Biobase_2.50.0
## [7] GenomicRanges_1.42.0 GenomeInfoDb_1.26.5
## [9] IRanges_2.24.1 S4Vectors_0.28.1
## [11] BiocGenerics_0.36.0 patchwork_1.1.1
## [13] ggplot2_3.3.3 SeuratObject_4.0.0
## [15] Seurat_4.0.1.9005 Signac_1.2.0
##
## loaded via a namespace (and not attached):
## [1] utf8_1.2.1 reticulate_1.19
## [3] tidyselect_1.1.0 RSQLite_2.2.7
## [5] htmlwidgets_1.5.3 grid_4.0.1
## [7] docopt_0.7.1 BiocParallel_1.24.1
## [9] Rtsne_0.15 munsell_0.5.0
## [11] codetools_0.2-18 ragg_1.1.2
## [13] ica_1.0-2 future_1.21.0
## [15] miniUI_0.1.1.1 withr_2.4.2
## [17] colorspace_2.0-0 highr_0.9
## [19] knitr_1.33 rstudioapi_0.13
## [21] ROCR_1.0-11 tensor_1.5
## [23] listenv_0.8.0 labeling_0.4.2
## [25] MatrixGenerics_1.2.1 slam_0.1-48
## [27] GenomeInfoDbData_1.2.4 polyclip_1.10-0
## [29] bit64_4.0.5 farver_2.1.0
## [31] rprojroot_2.0.2 parallelly_1.24.0
## [33] vctrs_0.3.7 generics_0.1.0
## [35] xfun_0.22 biovizBase_1.38.0
## [37] BiocFileCache_1.14.0 lsa_0.73.2
## [39] ggseqlogo_0.1 R6_2.5.0
## [41] hdf5r_1.3.3 DelayedArray_0.16.3
## [43] bitops_1.0-7 spatstat.utils_2.1-0
## [45] cachem_1.0.4 assertthat_0.2.1
## [47] promises_1.2.0.1 scales_1.1.1
## [49] nnet_7.3-15 gtable_0.3.0
## [51] globals_0.14.0 goftest_1.2-2
## [53] rlang_0.4.10 systemfonts_1.0.1
## [55] RcppRoll_0.3.0 splines_4.0.1
## [57] rtracklayer_1.50.0 lazyeval_0.2.2
## [59] dichromat_2.0-0 checkmate_2.0.0
## [61] spatstat.geom_2.1-0 yaml_2.2.1
## [63] reshape2_1.4.4 abind_1.4-5
## [65] backports_1.2.1 httpuv_1.6.0
## [67] Hmisc_4.5-0 tools_4.0.1
## [69] ellipsis_0.3.1 spatstat.core_2.1-2
## [71] jquerylib_0.1.4 RColorBrewer_1.1-2
## [73] ggridges_0.5.3 Rcpp_1.0.6
## [75] plyr_1.8.6 base64enc_0.1-3
## [77] progress_1.2.2 zlibbioc_1.36.0
## [79] purrr_0.3.4 RCurl_1.98-1.3
## [81] prettyunits_1.1.1 rpart_4.1-15
## [83] openssl_1.4.3 deldir_0.2-10
## [85] pbapply_1.4-3 cowplot_1.1.1
## [87] zoo_1.8-9 SummarizedExperiment_1.20.0
## [89] ggrepel_0.9.1 cluster_2.1.2
## [91] fs_1.5.0 magrittr_2.0.1
## [93] RSpectra_0.16-0 data.table_1.14.0
## [95] scattermore_0.7 lmtest_0.9-38
## [97] RANN_2.6.1 SnowballC_0.7.0
## [99] ProtGenerics_1.22.0 fitdistrplus_1.1-3
## [101] matrixStats_0.58.0 hms_1.0.0
## [103] mime_0.10 evaluate_0.14
## [105] xtable_1.8-4 XML_3.99-0.6
## [107] jpeg_0.1-8.1 sparsesvd_0.2
## [109] gridExtra_2.3 compiler_4.0.1
## [111] biomaRt_2.46.3 tibble_3.1.1
## [113] KernSmooth_2.23-18 crayon_1.4.1
## [115] htmltools_0.5.1.1 mgcv_1.8-33
## [117] later_1.2.0 Formula_1.2-4
## [119] tidyr_1.1.3 DBI_1.1.1
## [121] tweenr_1.0.2 dbplyr_2.1.1
## [123] MASS_7.3-53.1 rappdirs_0.3.3
## [125] Matrix_1.3-2 igraph_1.2.6
## [127] pkgconfig_2.0.3 pkgdown_1.6.1
## [129] GenomicAlignments_1.26.0 foreign_0.8-81
## [131] plotly_4.9.3 spatstat.sparse_2.0-0
## [133] xml2_1.3.2 bslib_0.2.4
## [135] XVector_0.30.0 VariantAnnotation_1.36.0
## [137] stringr_1.4.0 digest_0.6.27
## [139] sctransform_0.3.2 RcppAnnoy_0.0.18
## [141] spatstat.data_2.1-0 Biostrings_2.58.0
## [143] rmarkdown_2.7 leiden_0.3.7
## [145] fastmatch_1.1-0 htmlTable_2.1.0
## [147] uwot_0.1.10 curl_4.3
## [149] shiny_1.6.0 Rsamtools_2.6.0
## [151] lifecycle_1.0.0 nlme_3.1-152
## [153] jsonlite_1.7.2 BSgenome_1.58.0
## [155] desc_1.3.0 viridisLite_0.4.0
## [157] askpass_1.1 fansi_0.4.2
## [159] pillar_1.6.0 lattice_0.20-41
## [161] fastmap_1.1.0 httr_1.4.2
## [163] survival_3.2-11 glue_1.4.2
## [165] qlcMatrix_0.9.7 png_0.1-7
## [167] bit_4.0.4 ggforce_0.3.3
## [169] stringi_1.5.3 sass_0.3.1
## [171] blob_1.2.1 textshaping_0.3.3
## [173] latticeExtra_0.6-29 memoise_2.0.0
## [175] dplyr_1.0.5 irlba_2.3.3
## [177] future.apply_1.7.0