Analyzing PBMC scATAC-seq
Compiled: April 01, 2026
Source:vignettes/pbmc_vignette.Rmd
pbmc_vignette.RmdFor this tutorial, we will be analyzing a single-cell ATAC-seq dataset of human peripheral blood mononuclear cells (PBMCs) provided by 10x Genomics. The following files are used in this vignette, all available through the 10x Genomics website:
- The Raw
data
- The Metadata
- The fragments file
- The fragments file index
View data download code
To download all the required files, you can run the following lines in a shell:
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_filtered_peak_bc_matrix.h5
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_singlecell.csv
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz.tbiFirst load in Signac, Seurat, and some other packages we will be using for analyzing human data.
## Warning: package 'Seurat' was built under R version 4.5.2## Warning: package 'sp' was built under R version 4.5.2## Warning: package 'GenomicRanges' was built under R version 4.5.2## Warning: package 'ggplot2' was built under R version 4.5.2## Warning: package 'GenomeInfoDb' was built under R version 4.5.2Pre-processing workflow
When pre-processing chromatin data, Signac uses information from two related input files, both of which can be created using CellRanger:
Peak/Cell matrix. This is analogous to the gene expression count matrix used to analyze single-cell RNA-seq. However, instead of genes, each row of the matrix represents a region of the genome (a peak), that is predicted to represent a region of open chromatin. Each value in the matrix represents the number of Tn5 integration sites for each single barcode (i.e. a cell) that map within each peak. You can find more detail on the 10X Website.
Fragment file. This represents a full list of all unique fragments across all single cells. It is a substantially larger file, is slower to work with, and is stored on-disk (instead of in memory). However, the advantage of retaining this file is that it contains all fragments associated with each single cell, as opposed to only fragments that map to peaks. More information about the fragment file can be found on the 10x Genomics website or on the sinto website.
We start by creating a Seurat object using the peak/cell matrix and
cell metadata generated by cellranger-atac, and store the
path to the fragment file on disk in the Seurat object:
counts <- Read10X_h5(filename = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_filtered_peak_bc_matrix.h5")
metadata <- read.csv(
file = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_singlecell.csv",
header = TRUE,
row.names = 1
)
chrom_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
fragments = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz",
min.cells = 10,
min.features = 200
)
pbmc <- CreateSeuratObject(
counts = chrom_assay,
assay = "peaks",
meta.data = metadata
)
pbmc## An object of class Seurat
## 165434 features across 10246 samples within 1 assay
## Active assay: peaks (165434 features, 0 variable features)
## 2 layers present: counts, dataWhat if I don’t have an H5 file?
If you do not have the .h5 file, you can still run an
analysis. You may have data that is formatted as three files, a counts
file (.mtx), a cell barcodes file, and a peaks file. If
this is the case you can load the data using the
Matrix::readMM() function:
counts <- Matrix::readMM("filtered_peak_bc_matrix/matrix.mtx")
barcodes <- readLines("filtered_peak_bc_matrix/barcodes.tsv")
peaks <- read.table("filtered_peak_bc_matrix/peaks.bed", sep="\t")
peaknames <- paste(peaks$V1, peaks$V2, peaks$V3, sep="-")
colnames(counts) <- barcodes
rownames(counts) <- peaknamesAlternatively, you might only have a fragment file. In this case you
can create a count matrix using the FeatureMatrix()
function:
fragpath <- '10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz'
# Define cells
# If you already have a list of cell barcodes to use you can skip this step
total_counts <- CountFragments(fragpath)
cutoff <- 1000 # Change this number depending on your dataset!
barcodes <- total_counts[total_counts$frequency_count > cutoff, ]$CB
# Create a fragment object
frags <- CreateFragmentObject(path = fragpath, cells = barcodes)
# First call peaks on the dataset
# If you already have a set of peaks you can skip this step
peaks <- CallPeaks(frags)
# Quantify fragments in each peak
counts <- FeatureMatrix(fragments = frags, features = peaks, cells = barcodes)The ATAC-seq data is stored using a custom assay, the
ChromatinAssay. This enables some specialized functions for
analysing genomic single-cell assays such as scATAC-seq. By printing the
assay we can see some of the additional information that can be
contained in the ChromatinAssay, including motif
information, gene annotations, and genome information.
pbmc[['peaks']]## ChromatinAssay data with 165434 features for 10246 cells
## Variable features: 0
## Genome:
## Annotation present: FALSE
## Motifs present: FALSE
## Fragment files: 1For example, we can call granges on a Seurat object with
a ChromatinAssay set as the active assay (or on a
ChromatinAssay) to see the genomic ranges associated with
each feature in the object. See the object interaction vignette for more
information about the ChromatinAssay class.
granges(pbmc)## GRanges object with 165434 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 9772-10660 *
## [2] chr1 180712-181178 *
## [3] chr1 181200-181607 *
## [4] chr1 191183-192084 *
## [5] chr1 267576-268461 *
## ... ... ... ...
## [165430] KI270713.1 13054-13909 *
## [165431] KI270713.1 15212-15933 *
## [165432] KI270713.1 21459-22358 *
## [165433] KI270713.1 29676-30535 *
## [165434] KI270713.1 36913-37813 *
## -------
## seqinfo: 35 sequences from an unspecified genome; no seqlengthsWe then remove the features that correspond to chromosome scaffolds e.g. (KI270713.1) or other sequences instead of the (22+2) standard chromosomes.
peaks.keep <- seqnames(granges(pbmc)) %in% standardChromosomes(granges(pbmc))
pbmc <- pbmc[as.vector(peaks.keep), ]We can also add gene annotations to the pbmc object for
the human genome. This will allow downstream functions to pull the gene
annotation information directly from the object.
Multiple patches are released for each genome assembly. When dealing with mapped data (such as the 10x Genomics files we will be using), it is advisable to use the annotations from the same assembly patch that was used to perform the mapping.
From the dataset summary, we can see that the reference package 10x Genomics used to perform the mapping was “GRCh38-2020-A”, which corresponds to the Ensembl v98 patch release. For more information on the various Ensembl releases, you can refer to this site.
library(AnnotationHub)
ah <- AnnotationHub()
# Search for the Ensembl 98 EnsDb for Homo sapiens on AnnotationHub
query(ah, "EnsDb.Hsapiens.v98")## AnnotationHub with 1 record
## # snapshotDate(): 2025-10-29
## # names(): AH75011
## # $dataprovider: Ensembl
## # $species: Homo sapiens
## # $rdataclass: EnsDb
## # $rdatadateadded: 2019-05-02
## # $title: Ensembl 98 EnsDb for Homo sapiens
## # $description: Gene and protein annotations for Homo sapiens based on Ensembl version 98.
## # $taxonomyid: 9606
## # $genome: GRCh38
## # $sourcetype: ensembl
## # $sourceurl: http://www.ensembl.org
## # $sourcesize: NA
## # $tags: c("98", "AHEnsDbs", "Annotation", "EnsDb", "Ensembl", "Gene", "Protein", "Transcript")
## # retrieve record with 'object[["AH75011"]]'
ensdb_v98 <- ah[["AH75011"]]
# extract gene annotations from EnsDb
annotations <- GetGRangesFromEnsDb(ensdb = ensdb_v98)
# change to UCSC style since the data was mapped to hg38
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))
genome(annotations) <- "hg38"
# add the gene information to the object
Annotation(pbmc) <- annotationsComputing QC Metrics
We can now compute some QC metrics for the scATAC-seq experiment. We currently suggest the following metrics below to assess data quality. As with scRNA-seq, the expected range of values for these parameters will vary depending on your biological system, cell viability, and other factors.
Nucleosome banding pattern: The histogram of DNA fragment sizes (determined from the paired-end sequencing reads) should exhibit a strong nucleosome banding pattern corresponding to the length of DNA wrapped around a single nucleosome. We calculate this per single cell, and quantify the approximate ratio of mononucleosomal to nucleosome-free fragments (stored as
nucleosome_signal)Transcriptional start site (TSS) enrichment score: The ENCODE project has defined an ATAC-seq targeting score based on the ratio of fragments centered at the TSS to fragments in TSS-flanking regions (see https://www.encodeproject.org/data-standards/terms/). Poor ATAC-seq experiments typically will have a low TSS enrichment score. We can compute this metric for each cell with the
TSSEnrichment()function, and the results are stored in metadata under the column nameTSS.enrichment.Total number of fragments in peaks: A measure of cellular sequencing depth / complexity. Cells with very few reads may need to be excluded due to low sequencing depth. Cells with extremely high levels may represent doublets, nuclei clumps, or other artefacts.
Fraction of fragments in peaks: Represents the fraction of all fragments that fall within ATAC-seq peaks. Cells with low values (i.e. <15-20%) often represent low-quality cells or technical artifacts that should be removed. Note that this value can be sensitive to the set of peaks used.
Ratio reads in genomic blacklist regions: The ENCODE project has provided a list of blacklist regions, representing reads which are often associated with artefactual signal. Cells with a high proportion of reads mapping to these areas (compared to reads mapping to peaks) often represent technical artifacts and should be removed. The
FractionCountsInRegion()function can be used to calculate the fraction of all counts within a given set of regions per cell. We can use this function and a list of blacklist regions to find the fraction of blacklist counts per cell.
Note that the last three metrics can be obtained from the output of CellRanger (which is stored in the object metadata), but can also be calculated for non-10x datasets using Signac (more information at the end of this document).
# compute nucleosome signal score per cell
pbmc <- NucleosomeSignal(object = pbmc)
# compute TSS enrichment score per cell
pbmc <- TSSEnrichment(object = pbmc)
# add fraction of reads in peaks
pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100
# add blacklist ratio
blacklist_regions <- ah[['AH107305']] # blacklist regions for hg38
pbmc$blacklist_ratio <- FractionCountsInRegion(
object = pbmc,
assay = 'peaks',
regions = blacklist_regions
)The relationship between variables stored in the object metadata can
be visualized using the DensityScatter() function. This can
also be used to quickly find suitable cutoff values for different QC
metrics by setting quantiles=TRUE:
DensityScatter(pbmc, x = 'nCount_peaks', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)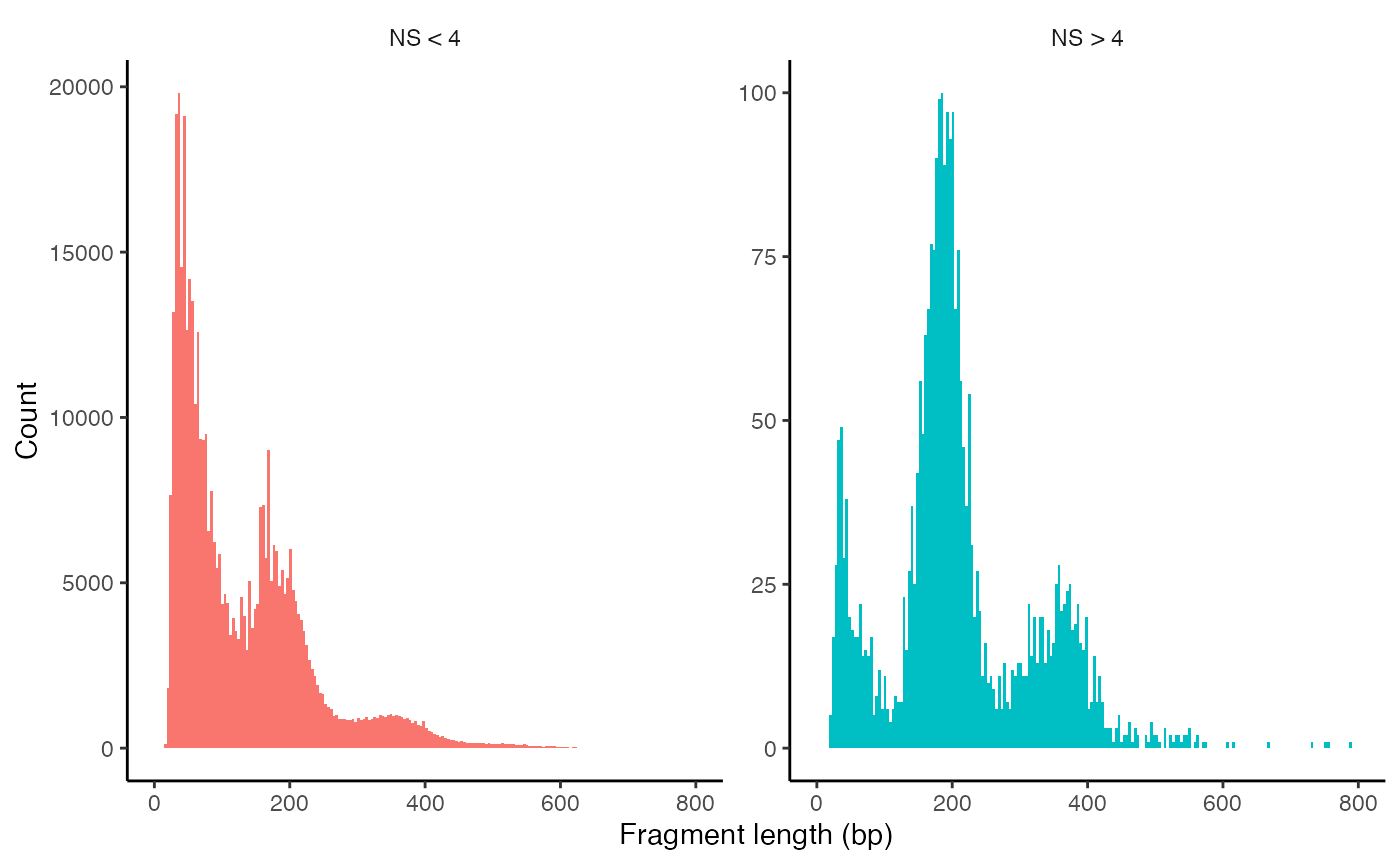
We can also look at the fragment length periodicity for all the cells, and group by cells with high or low nucleosomal signal strength. You can see that cells that are outliers for the mononucleosomal / nucleosome-free ratio (based on the plots above) have different nucleosomal banding patterns. The remaining cells exhibit a pattern that is typical for a successful ATAC-seq experiment.
pbmc$nucleosome_group <- ifelse(pbmc$nucleosome_signal > 4, 'NS > 4', 'NS < 4')
FragmentHistogram(object = pbmc, group.by = 'nucleosome_group')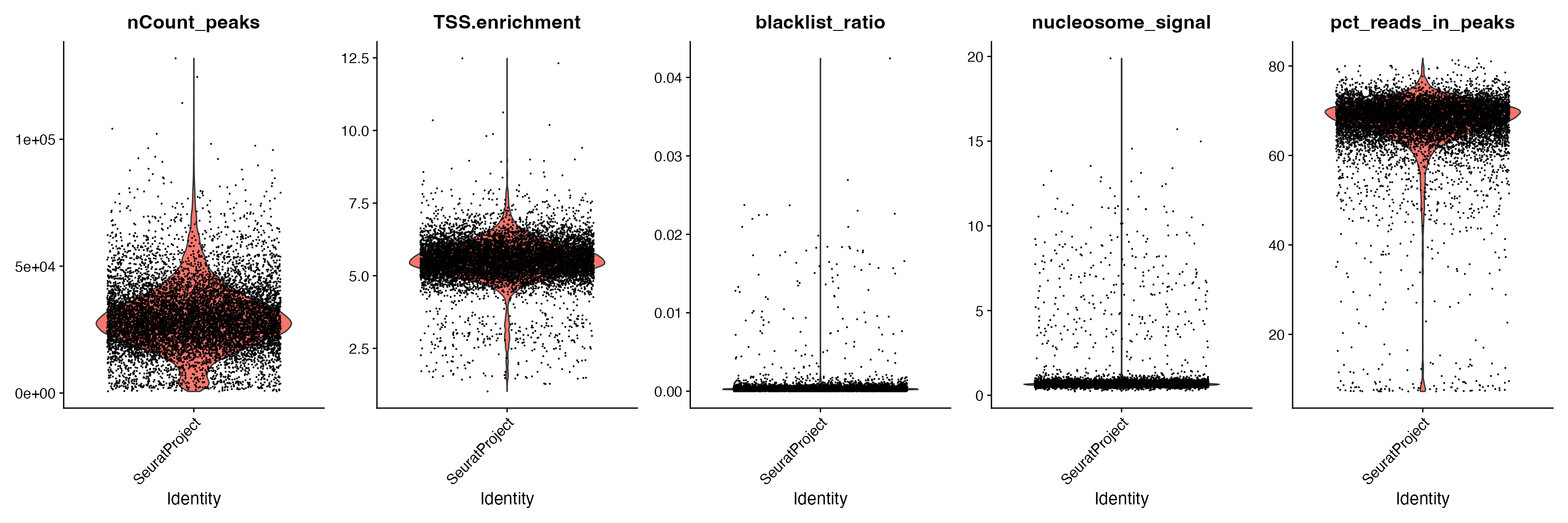
We can plot the distribution of each QC metric separately using a violin plot:
VlnPlot(
object = pbmc,
features = c('nCount_peaks', 'TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal', 'pct_reads_in_peaks'),
pt.size = 0.1,
ncol = 5
)
Finally we remove cells that are outliers for these QC metrics. The exact QC thresholds used will need to be adjusted according to your dataset.
pbmc <- subset(
x = pbmc,
subset = nCount_peaks > 9000 &
nCount_peaks < 100000 &
pct_reads_in_peaks > 40 &
blacklist_ratio < 0.01 &
nucleosome_signal < 4 &
TSS.enrichment > 4
)
pbmc## An object of class Seurat
## 165376 features across 9651 samples within 1 assay
## Active assay: peaks (165376 features, 0 variable features)
## 2 layers present: counts, dataNormalization and linear dimensional reduction
Normalization: Signac performs term frequency-inverse document frequency (TF-IDF) normalization. This is a two-step normalization procedure, that both normalizes across cells to correct for differences in cellular sequencing depth, and across peaks to give higher values to more rare peaks.
Feature selection: The low dynamic range of scATAC-seq data makes it challenging to perform variable feature selection, as we do for scRNA-seq. Instead, we can choose to use only the top n% of features (peaks) for dimensional reduction, or remove features present in less than n cells with the
FindTopFeatures()function. Here we will use all features, though we have seen very similar results when using only a subset of features (try setting min.cutoff to ‘q75’ to use the top 25% all peaks), with faster runtimes. Features used for dimensional reduction are automatically set asVariableFeatures()for the Seurat object by this function.Dimension reduction: We next run singular value decomposition (SVD) on the TD-IDF matrix, using the features (peaks) selected above. This returns a reduced dimension representation of the object (for users who are more familiar with scRNA-seq, you can think of this as analogous to the output of PCA).
The combined steps of TF-IDF followed by SVD are known as latent semantic indexing (LSI), and were first introduced for the analysis of scATAC-seq data by Cusanovich et al. 2015.
pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
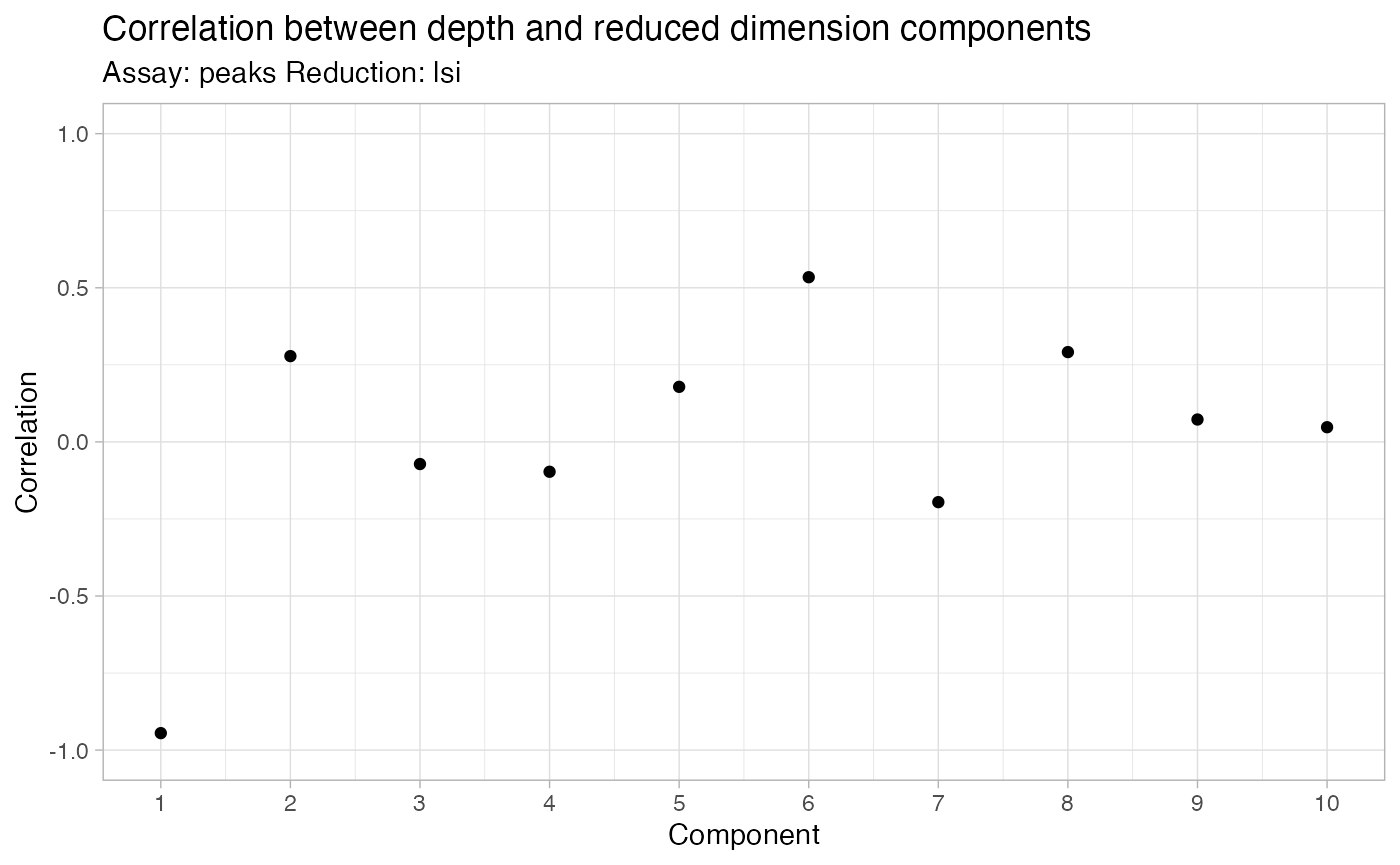
pbmc <- RunSVD(pbmc)The first LSI component often captures sequencing depth (technical
variation) rather than biological variation. If this is the case, the
component should be removed from downstream analysis. We can assess the
correlation between each LSI component and sequencing depth using the
DepthCor() function:
DepthCor(pbmc)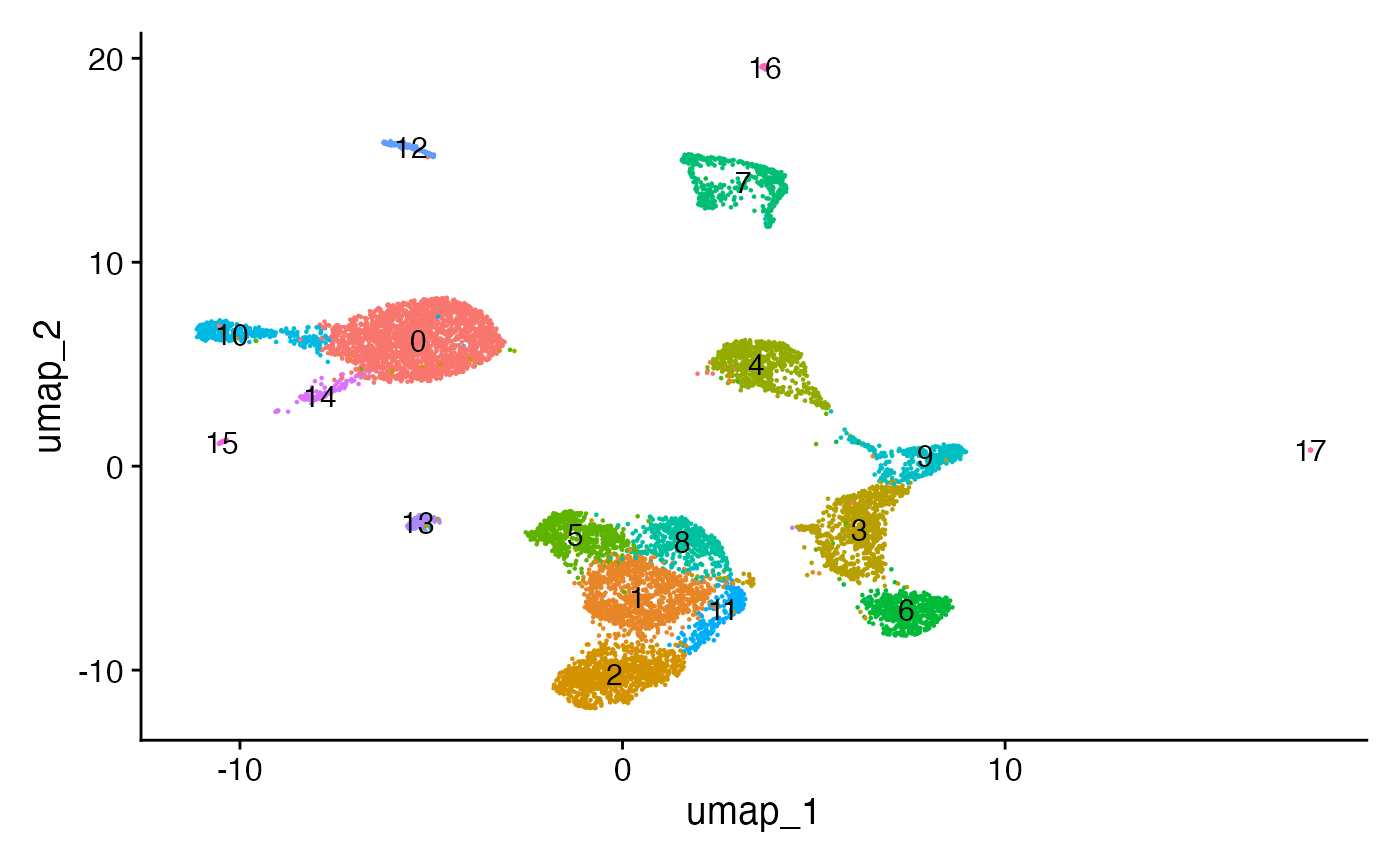
Here we see there is a very strong correlation between the first LSI component and the total number of counts for the cell. We will perform downstream steps without this component as we don’t want to group cells together based on their total sequencing depth, but rather by their patterns of accessibility at cell-type-specific peaks.
Non-linear dimension reduction and clustering
Now that the cells are embedded in a low-dimensional space we can use
methods commonly applied for the analysis of scRNA-seq data to perform
graph-based clustering and non-linear dimension reduction for
visualization. The functions RunUMAP(),
FindNeighbors(), and FindClusters() all come
from the Seurat package.
pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = 2:30)
pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = 2:30)
pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)
DimPlot(object = pbmc, label = TRUE) + NoLegend()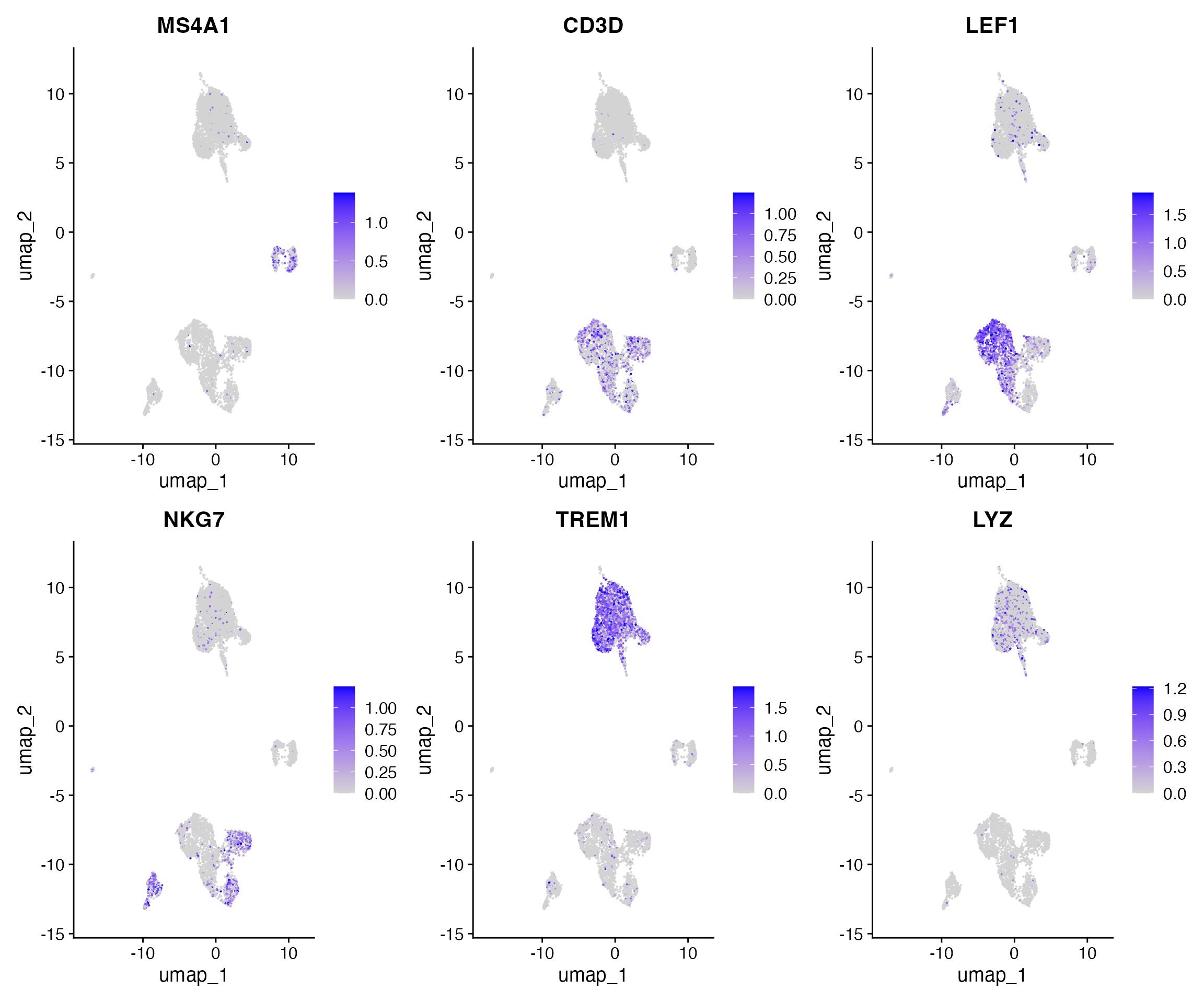
Create a gene activity matrix
The UMAP visualization reveals the presence of multiple cell groups in human blood. If you are familiar with scRNA-seq analyses of PBMC, you may recognize the presence of certain myeloid and lymphoid populations in the scATAC-seq data. However, annotating and interpreting clusters is more challenging in scATAC-seq data as much less is known about the functional roles of noncoding genomic regions than is known about protein coding regions (genes).
We can try to quantify the activity of each gene in the genome by assessing the chromatin accessibility associated with the gene, and create a new gene activity assay derived from the scATAC-seq data. Here we will use a simple approach of summing the fragments intersecting the gene body and promoter region (we also recommend exploring the Cicero tool, which can accomplish a similar goal, and we provide a vignette showing how to run Cicero within a Signac workflow here).
To create a gene activity matrix, we extract gene coordinates and
extend them to include the 2 kb upstream region (as promoter
accessibility is often correlated with gene expression). We then count
the number of fragments for each cell that map to each of these regions,
using the using the FeatureMatrix() function. These steps
are automatically performed by the GeneActivity()
function:
gene.activities <- GeneActivity(pbmc)
# add the gene activity matrix to the Seurat object as a new assay and normalize it
pbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)
pbmc <- NormalizeData(
object = pbmc,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(pbmc$nCount_RNA)
)Now we can visualize the activities of canonical marker genes to help interpret our ATAC-seq clusters. Note that the activities will be much noisier than scRNA-seq measurements. This is because they represent measurements from sparse chromatin data, and because they assume a general correspondence between gene body/promoter accessibility and gene expression which may not always be the case. Nonetheless, we can begin to discern populations of monocytes, B, T, and NK cells based on these gene activity profiles. However, further subdivision of these cell types is challenging based on supervised analysis alone.
DefaultAssay(pbmc) <- 'RNA'
FeaturePlot(
object = pbmc,
features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 3
)
Integrating with scRNA-seq data
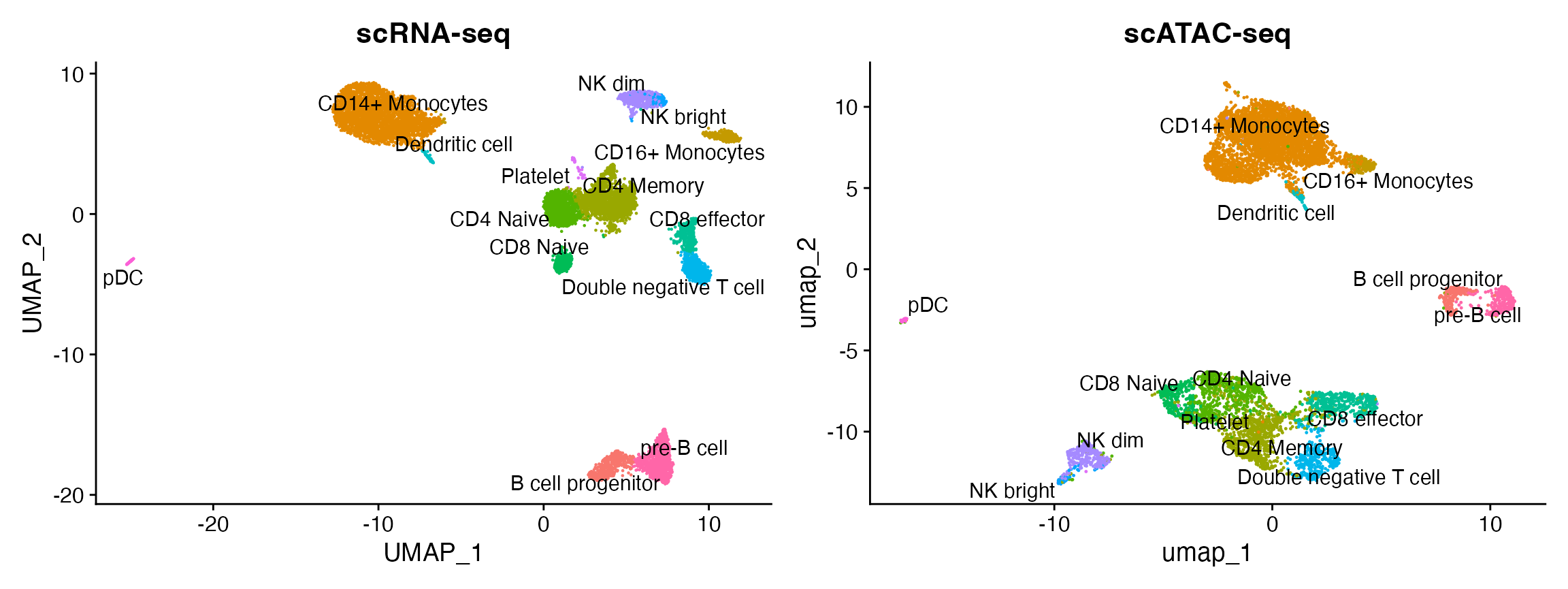
To help interpret the scATAC-seq data, we can classify cells based on an scRNA-seq experiment from the same biological system (human PBMC). We utilize methods for cross-modality integration and label transfer, described here, with a more in-depth tutorial here. We aim to identify shared correlation patterns in the gene activity matrix and scRNA-seq dataset to identify matched biological states across the two modalities. This procedure returns a classification score for each cell for each scRNA-seq-defined cluster label.
Here we load a pre-processed scRNA-seq dataset for human PBMCs, also provided by 10x Genomics. You can download the raw data for this experiment from the 10x website, and view the code used to construct this object on GitHub. Alternatively, you can download the pre-processed Seurat object here.
# Load the pre-processed scRNA-seq data for PBMCs
pbmc_rna <- readRDS("pbmc_10k_v3.rds")
pbmc_rna <- UpdateSeuratObject(pbmc_rna)
transfer.anchors <- FindTransferAnchors(
reference = pbmc_rna,
query = pbmc,
reduction = 'cca'
)## Running CCA## Merging objects## Finding neighborhoods## Finding anchors## Found 19096 anchors
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = pbmc_rna$celltype,
weight.reduction = pbmc[['lsi']],
dims = 2:30
)## Finding integration vectors## Finding integration vector weights## Predicting cell labels
pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
plot1 <- DimPlot(
object = pbmc_rna,
group.by = 'celltype',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scRNA-seq')
plot2 <- DimPlot(
object = pbmc,
group.by = 'predicted.id',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')
plot1 + plot2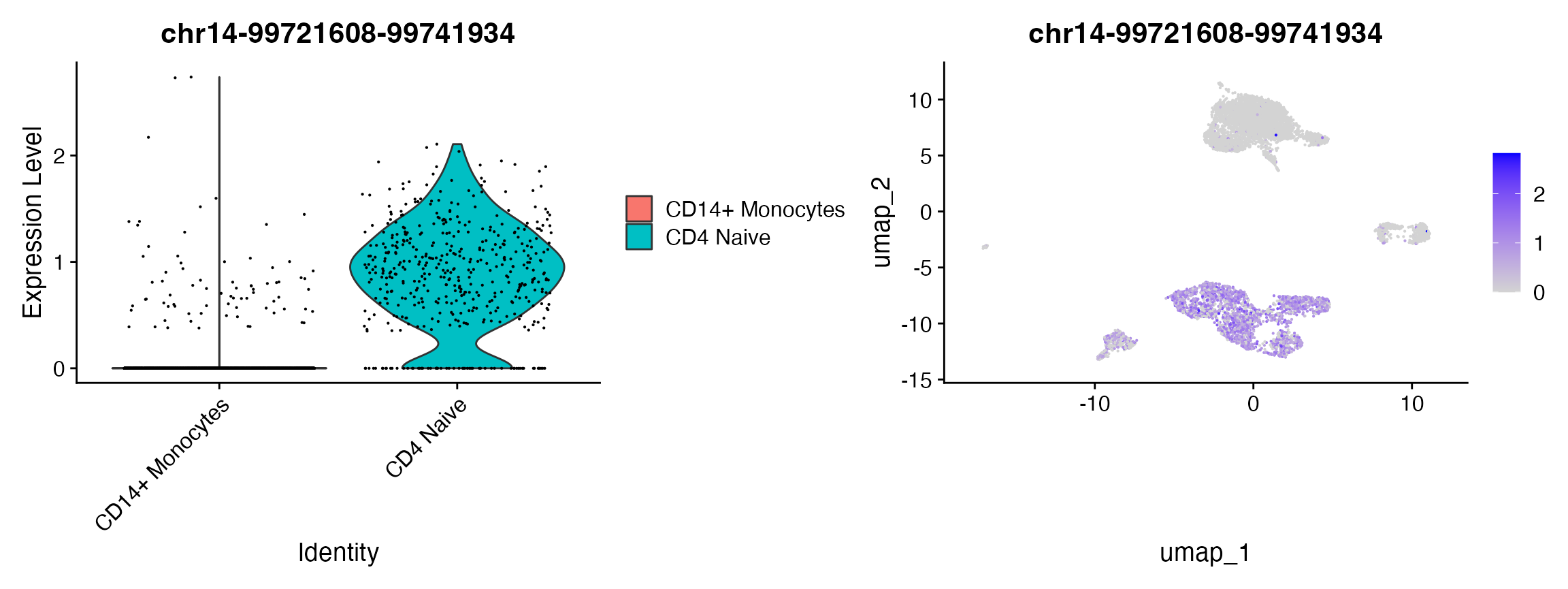
You can see that the scRNA-based classifications are consistent with the UMAP visualization that was computed using the scATAC-seq data. Notice, however, that a small population of cells are predicted to be platelets in the scATAC-seq dataset. This is unexpected as platelets are not nucleated and should not be detected by scATAC-seq. It is possible that the cells predicted to be platelets could instead be the platelet-precursors megakaryocytes, which largely reside in the bone marrow but are rarely found in the peripheral blood of healthy patients, such as the individual these PBMCs were drawn from. Given the already extreme rarity of megakaryocytes within normal bone marrow (< 0.1%), this scenario seems unlikely.
A closer look at the “platelets”
Plotting the prediction score for the cells assigned to each label reveals that the “platelet” cells received relatively low scores (< 0.8), indicating a low confidence in the assigned cell identity. In most cases, the next most likely cell identity predicted for these cells was “CD4 naive”.
VlnPlot(pbmc, 'prediction.score.max', group.by = 'predicted.id')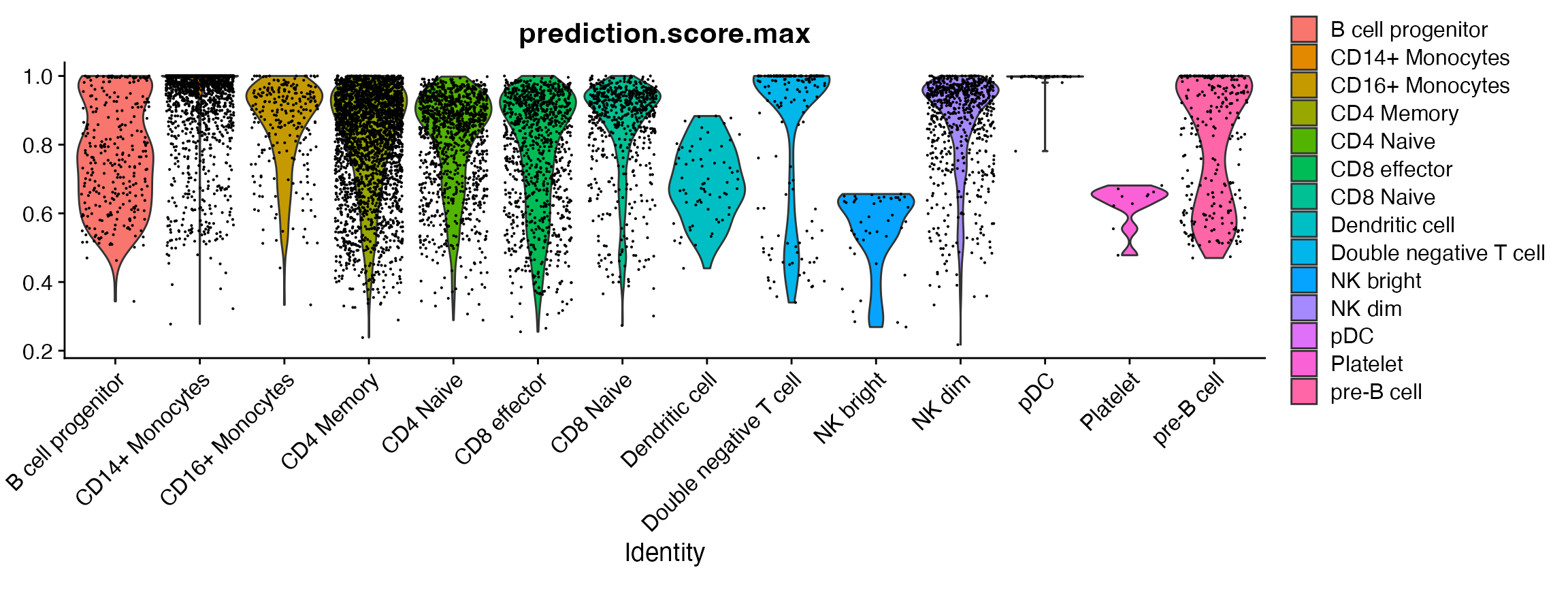
# Identify the metadata columns that start with "prediction.score."
metadata_attributes <- colnames(pbmc[[]])
prediction_score_attributes <- grep("^prediction.score.", metadata_attributes, value = TRUE)
prediction_score_attributes <- setdiff(prediction_score_attributes, "prediction.score.max")
# Extract the prediction score attributes for these cells
predicted_platelets <- which(pbmc$predicted.id == "Platelet")
platelet_scores <- pbmc[[]][predicted_platelets, prediction_score_attributes]
# Order the columns by their average values in descending order
ordered_columns <- names(sort(colMeans(platelet_scores, na.rm = TRUE), decreasing = TRUE))
ordered_platelet_scores_df <- platelet_scores[, ordered_columns]
print(ordered_platelet_scores_df)## prediction.score.Platelet prediction.score.CD4.Naive prediction.score.CD4.Memory prediction.score.pre.B.cell prediction.score.CD14..Monocytes prediction.score.NK.dim prediction.score.CD8.Naive prediction.score.CD8.effector prediction.score.Double.negative.T.cell prediction.score.NK.bright prediction.score.pDC prediction.score.CD16..Monocytes prediction.score.B.cell.progenitor prediction.score.Dendritic.cell
## ACAAAGAAGACACGGT-1 0.6223878 0.319437652 0.03441178 0.02179066 0.001654916 0.0000000 0.00000000 0.0003171606 0.00000000 0 0 0 0 0
## CACTAAGGTAATGTAG-1 0.4922400 0.208920020 0.02069596 0.05901881 0.065733566 0.1123977 0.03349192 0.0075020529 0.00000000 0 0 0 0 0
## CTCAACCAGCGAGCTA-1 0.6496477 0.289615288 0.03165959 0.01812907 0.010948402 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## GAATCTGCATAGTCCA-1 0.6667046 0.273470367 0.02347730 0.01696178 0.019385979 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## GCTTAAGCAAAGGTCG-1 0.6608942 0.266923116 0.02732163 0.01932498 0.025536052 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## GTCACCTGTCAGGCTC-1 0.6488676 0.280942136 0.03177828 0.02565093 0.012761077 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## TAATCGGTCGTAGCGC-1 0.6791654 0.258052454 0.02748286 0.02293892 0.012360320 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## TACTAGGGTATGGATA-1 0.6533584 0.284576943 0.03000663 0.02148336 0.010574668 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0
## TCCAGAAGTAGCGTTT-1 0.4779871 0.004982414 0.38042596 0.01261527 0.000000000 0.0000000 0.05246751 0.0338928296 0.03762892 0 0 0 0 0
## TCGATTTGTGTGTGCC-1 0.6290691 0.301177637 0.04006119 0.01985544 0.009836580 0.0000000 0.00000000 0.0000000000 0.00000000 0 0 0 0 0As there are only a very small number of cells classified as “platelets” (< 20), it is difficult to figure out their precise cellular identity. Larger datasets would be required to confidently identify specific peaks for this population of cells, and further analysis performed to correctly annotate them. For downstream analysis we will thus remove the extremely rare cell states that were predicted, retaining only cell annotations with >20 cells total.
predicted_id_counts <- table(pbmc$predicted.id)
# Identify the predicted.id values that have more than 20 cells
major_predicted_ids <- names(predicted_id_counts[predicted_id_counts > 20])
pbmc <- pbmc[, pbmc$predicted.id %in% major_predicted_ids]For downstream analyses, we can simply reassign the identities of each cell from their UMAP cluster index to the per-cell predicted labels. It is also possible to consider merging the cluster indexes and predicted labels.
# change cell identities to the per-cell predicted labels
Idents(pbmc) <- pbmc$predicted.idTo combine clustering and label transfer results
Alternatively, in instances where we wish to combine our scATAC-seq clustering and label transfer results, we can reassign the identities of all cells within each UMAP cluster to the most common predicted label for that cluster.
Find differentially accessible peaks between cell types
To find differentially accessible regions between clusters of cells, we can perform a differential accessibility (DA) test. A simple approach is to perform a Wilcoxon rank sum test, and the presto package has implemented an extremely fast Wilcoxon test that can be run on a Seurat object.
Install presto
if (!requireNamespace("remotes", quietly = TRUE))
install.packages('remotes')
remotes::install_github('immunogenomics/presto')Here we will focus on comparing Naive CD4 cells and CD14 monocytes, but any groups of cells can be compared using these methods. We can also visualize these marker peaks on a violin plot, feature plot, dot plot, heat map, or any visualization tool in Seurat.
# change back to working with peaks instead of gene activities
DefaultAssay(pbmc) <- 'peaks'
# wilcox is the default option for test.use
da_peaks <- FindMarkers(
object = pbmc,
ident.1 = "CD4 Naive",
ident.2 = "CD14+ Monocytes",
test.use = 'wilcox',
min.pct = 0.1
)
head(da_peaks)## p_val avg_log2FC pct.1 pct.2 p_val_adj
## chr12-119988511-119989430 0 4.141853 0.782 0.090 0
## chr7-142808530-142809435 0 3.650239 0.758 0.088 0
## chr17-82126458-82127377 0 4.991398 0.705 0.043 0
## chr7-138752283-138753197 0 4.873302 0.685 0.046 0
## chr11-60985909-60986801 0 4.522416 0.659 0.062 0
## chr4-152100248-152101142 0 4.994737 0.622 0.038 0Logistic regression with total fragment number as the latent variable
Another approach for differential testing is to utilize logistic regression for, as suggested by Ntranos et al. 2018 for scRNA-seq data, and add the total number of fragments as a latent variable to mitigate the effect of differential sequencing depth on the result.
# change back to working with peaks instead of gene activities
DefaultAssay(pbmc) <- 'peaks'
# Use logistic regression, set total fragment no. as latent var
lr_da_peaks <- FindMarkers(
object = pbmc,
ident.1 = "CD4 Naive",
ident.2 = "CD14+ Monocytes",
test.use = 'LR',
latent.vars = 'nCount_peaks',
min.pct = 0.1
)
head(lr_da_peaks)
plot1 <- VlnPlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1,
idents = c("CD4 Naive","CD14+ Monocytes")
)
plot2 <- FeaturePlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1
)
plot1 | plot2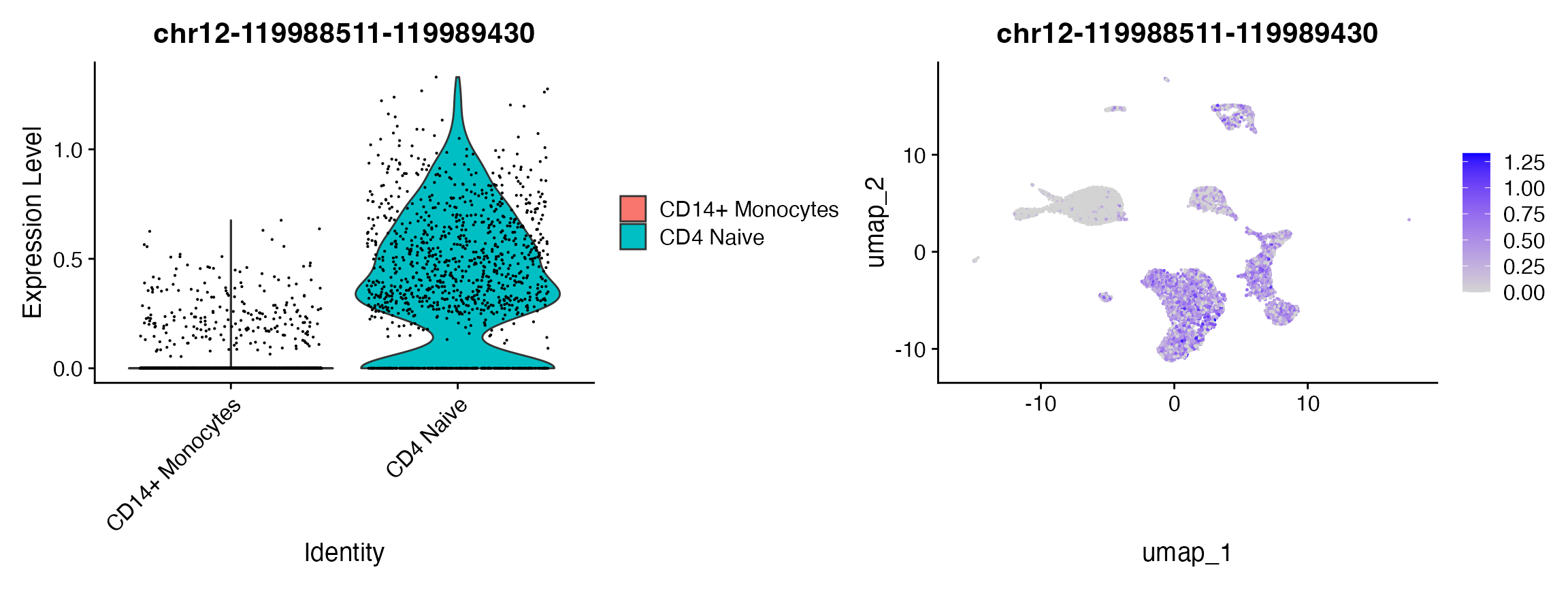
Peak coordinates can be difficult to interpret alone. We can find the
closest gene to each of these peaks using the
ClosestFeature() function.
open_cd4naive <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
open_cd14mono <- rownames(da_peaks[da_peaks$avg_log2FC < -3, ])
closest_genes_cd4naive <- ClosestFeature(pbmc, regions = open_cd4naive)
closest_genes_cd14mono <- ClosestFeature(pbmc, regions = open_cd14mono)
head(closest_genes_cd4naive)## tx_id gene_name gene_id gene_biotype type closest_region query_region distance
## ENSE00002206071 ENST00000397558 BICDL1 ENSG00000135127 protein_coding exon chr12-119989869-119990297 chr12-119988511-119989430 438
## ENST00000632998 ENST00000632998 PRSS2 ENSG00000275896 protein_coding utr chr7-142774509-142774564 chr7-142808530-142809435 33965
## ENST00000665763 ENST00000665763 CCDC57 ENSG00000176155 protein_coding gap chr17-82101867-82127691 chr17-82126458-82127377 0
## ENST00000645515 ENST00000645515 ATP6V0A4 ENSG00000105929 protein_coding cds chr7-138752625-138752837 chr7-138752283-138753197 0
## ENST00000545320 ENST00000545320 CD6 ENSG00000013725 protein_coding gap chr11-60971915-60987907 chr11-60985909-60986801 0
## ENST00000603548 ENST00000603548 FBXW7 ENSG00000109670 protein_coding utr chr4-152320544-152322880 chr4-152100248-152101142 219401
head(closest_genes_cd14mono)## tx_id gene_name gene_id gene_biotype type closest_region query_region distance
## ENST00000606214 ENST00000606214 TBC1D7 ENSG00000145979 protein_coding gap chr6-13267836-13305061 chr6-13302533-13303459 0
## ENST00000448962 ENST00000448962 RPS9 ENSG00000170889 protein_coding gap chr19-54201610-54231740 chr19-54207815-54208728 0
## ENST00000592988 ENST00000592988 AFMID ENSG00000183077 protein_coding gap chr17-78191061-78202498 chr17-78198651-78199583 0
## ENST00000336600 ENST00000336600 C6orf223 ENSG00000181577 protein_coding utr chr6-44003127-44007612 chr6-44058439-44059230 50826
## ENSE00002618192 ENST00000569518 VCPKMT ENSG00000100483 protein_coding exon chr14-50108632-50109713 chr14-50038381-50039286 69345
## ENSE00001389095 ENST00000340607 PTGES ENSG00000148344 protein_coding exon chr9-129752887-129753042 chr9-129776928-129777838 23885We could follow up with this result by doing gene ontology enrichment
analysis on the gene sets returned by ClosestFeature(),and
there are many R packages that can do this (see theGOstats
or clusterProfiler
packages for example).
GO enrichment analysis with clusterProfiler
## Warning: package 'clusterProfiler' was built under R version 4.5.2
library(org.Hs.eg.db)
library(enrichplot)## Warning: package 'enrichplot' was built under R version 4.5.2
cd4naive_ego <- enrichGO(gene = closest_genes_cd4naive$gene_id,
keyType = "ENSEMBL",
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
barplot(cd4naive_ego,showCategory = 20)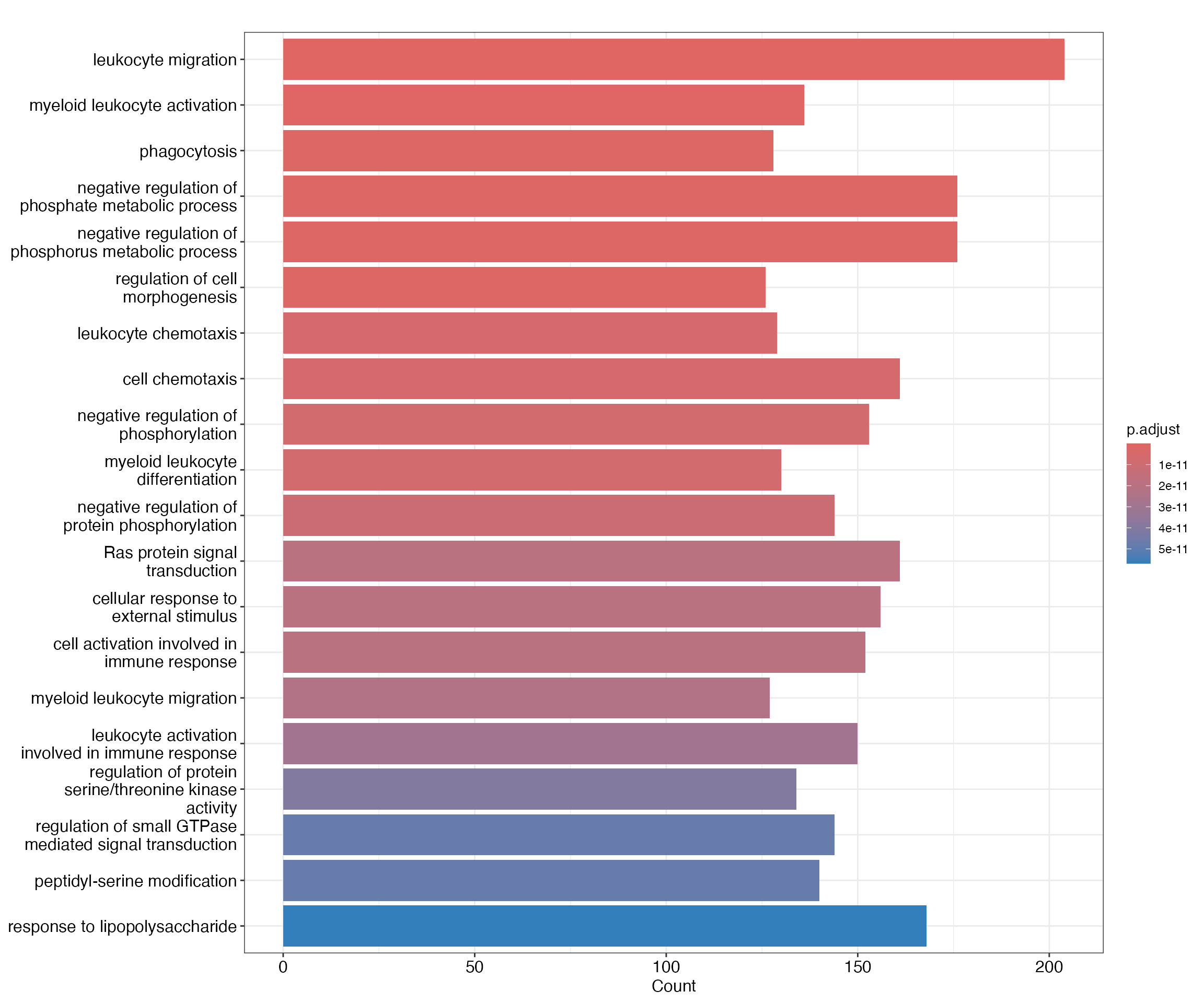
cd14mono_ego <- enrichGO(gene = closest_genes_cd14mono$gene_id,
keyType = "ENSEMBL",
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
barplot(cd14mono_ego,showCategory = 20)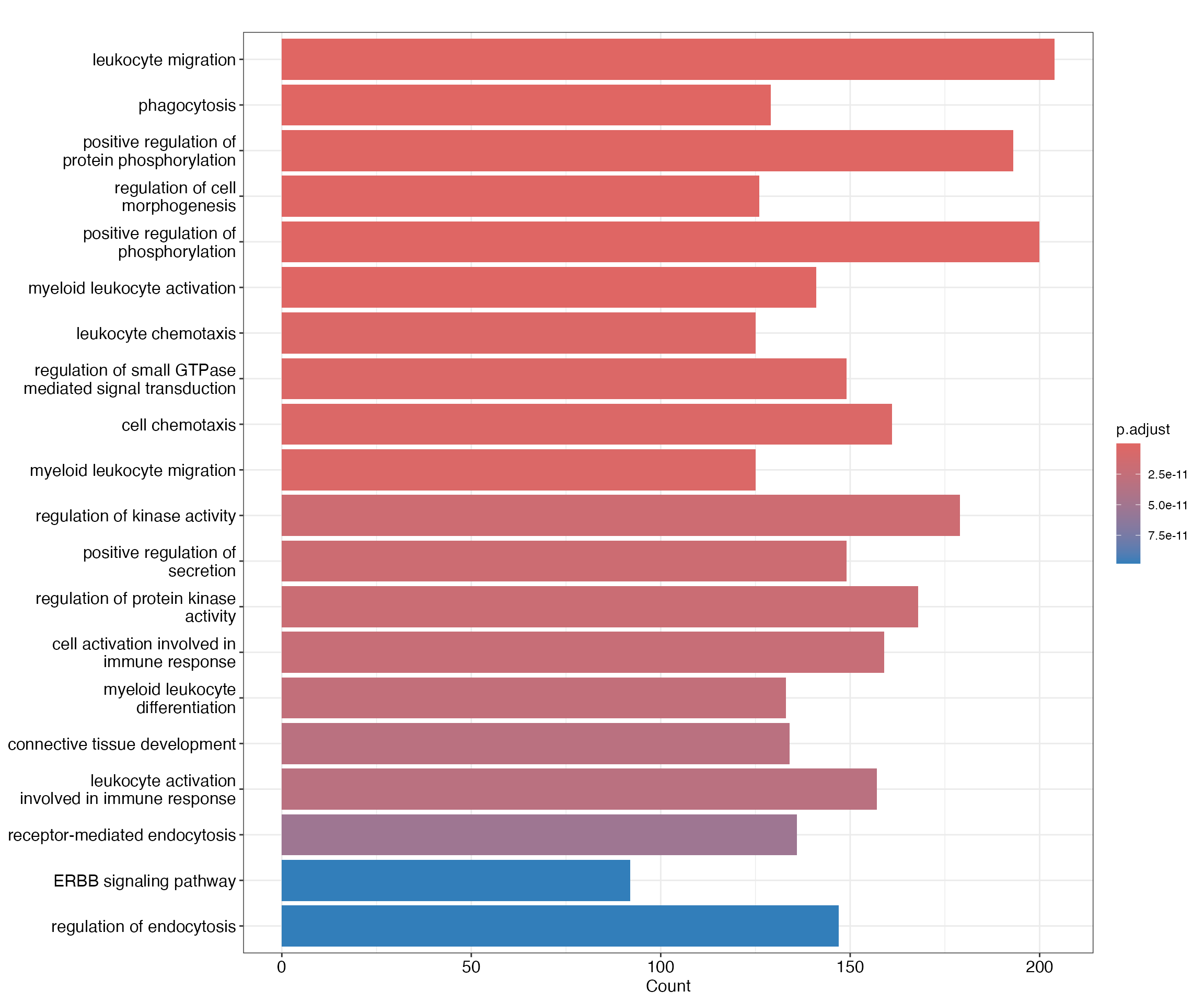
Plotting genomic regions
We can plot the frequency of Tn5 integration across regions of the
genome for cells grouped by cluster, cell type, or any other metadata
stored in the object for any genomic region using the
CoveragePlot() function. These represent pseudo-bulk
accessibility tracks, where signal from all cells within a group have
been averaged together to visualize the DNA accessibility in a region
(thanks to Andrew Hill for giving the inspiration for this function in
his excellent blog
post). Alongside these accessibility tracks we can visualize other
important information including gene annotation, peak coordinates, and
genomic links (if they’re stored in the object). See the visualization vignette for more
information.
For plotting purposes, it’s nice to have related cell types grouped
together. We can automatically sort the plotting order according to
similarities across the annotated cell types by running the
SortIdents() function:
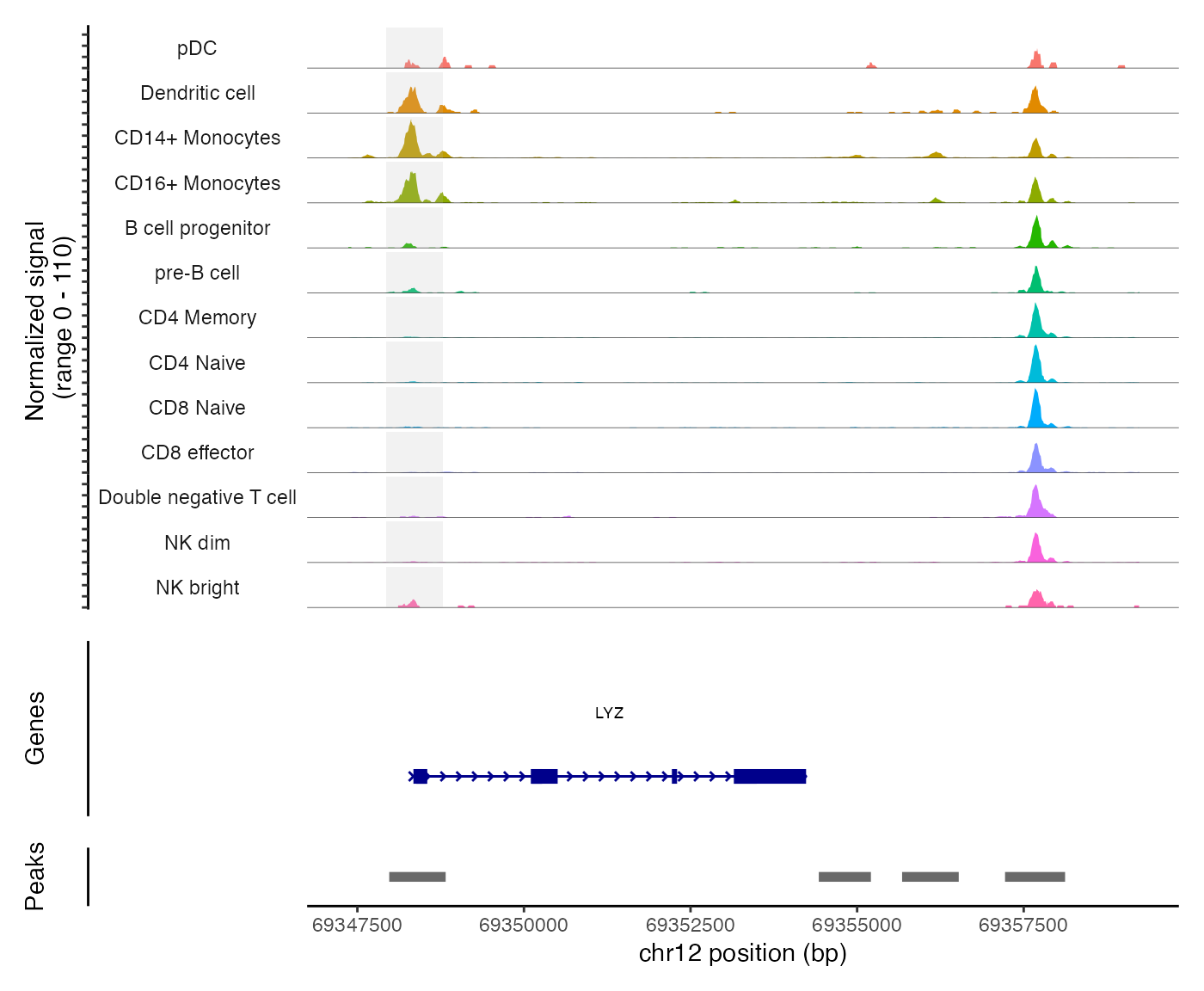
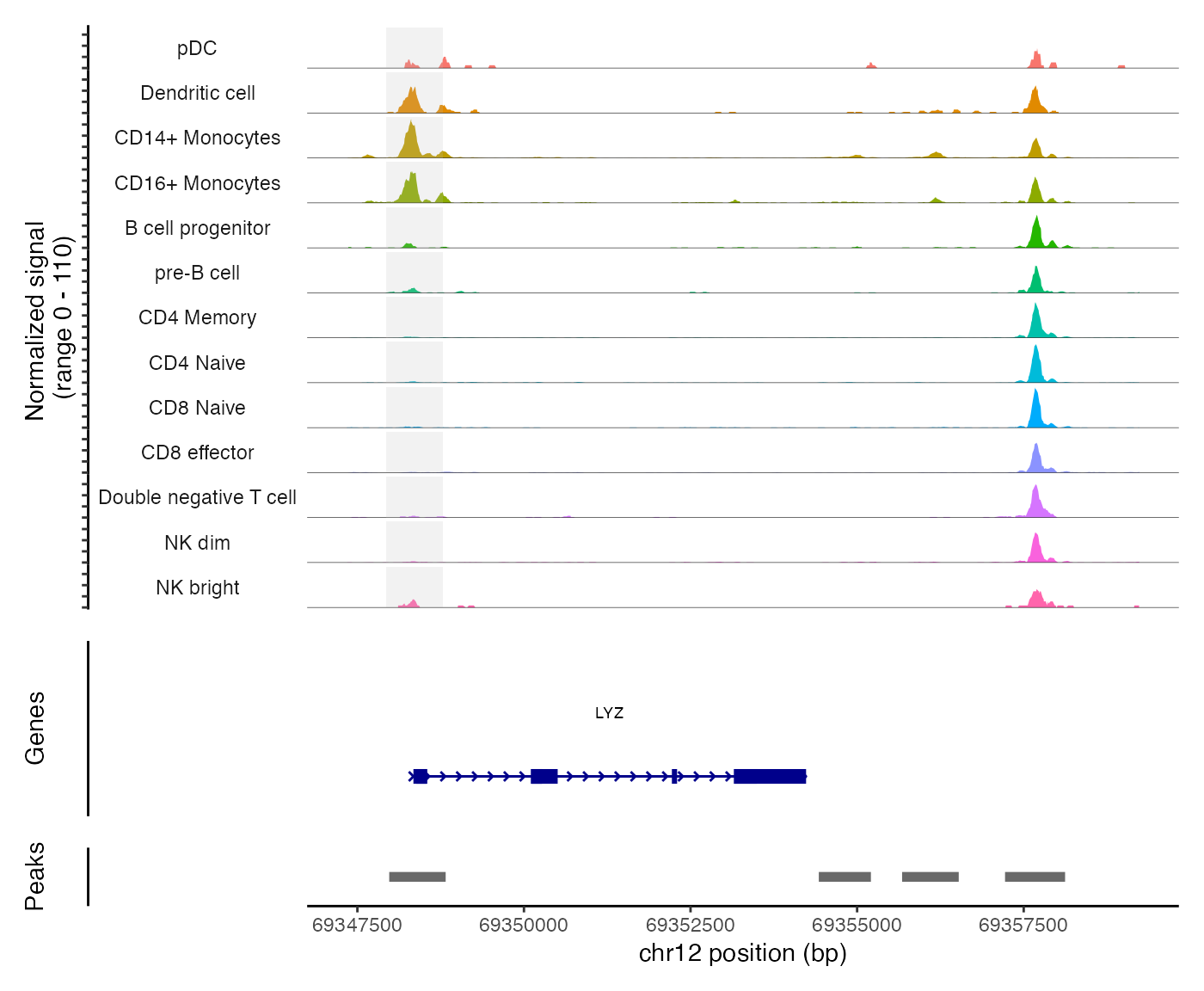
pbmc <- SortIdents(pbmc)We can then visualize the DA peaks open in CD4 naive cells and CD14 monocytes, near some key marker genes for these cell types, CD4 and LYZ respectively. Here we’ll highlight the DA peaks regions in grey.
# find DA peaks overlapping gene of interest
regions_highlight <- subsetByOverlaps(StringToGRanges(open_cd4naive), LookupGeneCoords(pbmc, "CD4"))
CoveragePlot(
object = pbmc,
region = "CD4",
region.highlight = regions_highlight,
extend.upstream = 1000,
extend.downstream = 1000
)
regions_highlight <- subsetByOverlaps(StringToGRanges(open_cd14mono), LookupGeneCoords(pbmc, "LYZ"))
CoveragePlot(
object = pbmc,
region = "LYZ",
region.highlight = regions_highlight,
extend.upstream = 1000,
extend.downstream = 5000
)
We can also create an interactive version of these plots using the
CoverageBrowser() function. Here is a recorded
demonstration showing how we can use CoverageBrowser() to
browser the genome and adjust plotting parameters interactively. The
“Save plot” button in CoverageBrowser() will add the
current plot to a list of ggplot objects that is returned
when the browser session is ended by pressing the “Done” button,
allowing interesting views to be saved during an interactive
session.
Working with datasets that were not quantified using CellRanger
The CellRanger software from 10x Genomics generates several useful QC metrics per-cell, as well as a peak/cell matrix and an indexed fragments file. In the above vignette, we utilize the CellRanger outputs, but provide alternative functions in Signac for many of the same purposes here.
Generating a peak/cell or bin/cell matrix
The FeatureMatrix function can be used to generate a
count matrix containing any set of genomic ranges in its rows. These
regions could be a set of peaks, or bins that span the entire
genome.
# not run
# peak_ranges should be a set of genomic ranges spanning the set of peaks to be quantified per cell
peak_matrix <- FeatureMatrix(
fragments = Fragments(pbmc),
features = peak_ranges
)For convenience, we also include a GenomeBinMatrix()
function that will generate a set of genomic ranges spanning the entire
genome for you, and run FeatureMatrix() internally to
produce a genome bin/cell matrix.
# not run
bin_matrix <- GenomeBinMatrix(
fragments = Fragments(pbmc),
genome = seqlengths(pbmc),
binsize = 5000
)Counting fraction of reads in peaks
The function FRiP() will count the fraction of reads in
peaks for each cell, given a peak/cell assay and a bin/cell assay. Note
that this can be run on a subset of the genome, so that a bin/cell assay
does not need to be computed for the whole genome. This will return a
Seurat object will metadata added corresponding to the fraction of reads
in peaks for each cell.
# not run
total_fragments <- CountFragments('10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz')
rownames(total_fragments) <- total_fragments$CB
pbmc$fragments <- total_fragments[colnames(pbmc), "frequency_count"]
pbmc <- FRiP(
object = pbmc,
assay = 'peaks',
total.fragments = 'fragments'
)Counting fragments in genome blacklist regions
The ratio of reads in genomic blacklist regions, that are known to
artifactually accumulate reads in genome sequencing assays, can be
diagnostic of low-quality cells. Genomic blacklist regions for a variety
of species can be found in the excluderanges
package available through AnnotationHub.
The FractionCountsInRegion() function can be used to
calculate the fraction of all counts within a given set of regions per
cell. We can use this function and the blacklist regions to find the
fraction of blacklist counts per cell.
# not run
pbmc$blacklist_fraction <- FractionCountsInRegion(
object = pbmc,
assay = 'peaks',
regions = blacklist_regions
)Session Info
## R version 4.5.1 (2025-06-13)
## Platform: aarch64-apple-darwin20
## Running under: macOS Tahoe 26.4
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: Asia/Singapore
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] enrichplot_1.30.4 org.Hs.eg.db_3.22.0 clusterProfiler_4.18.4 future_1.70.0 ensembldb_2.34.0 AnnotationFilter_1.34.0 GenomicFeatures_1.62.0 AnnotationDbi_1.72.0 Biobase_2.70.0 AnnotationHub_4.0.0 BiocFileCache_3.0.0 dbplyr_2.5.2 GenomeInfoDb_1.46.2 patchwork_1.3.2 ggplot2_4.0.2 GenomicRanges_1.62.1 Seqinfo_1.0.0 IRanges_2.44.0 S4Vectors_0.48.0 BiocGenerics_0.56.0 generics_0.1.4 Seurat_5.4.0 SeuratObject_5.3.0.9002 sp_2.2-1
## [25] Signac_1.17.0
##
## loaded via a namespace (and not attached):
## [1] fs_2.0.1 ProtGenerics_1.42.0 matrixStats_1.5.0 spatstat.sparse_3.1-0 bitops_1.0-9 httr_1.4.8 RColorBrewer_1.1-3 tools_4.5.1 sctransform_0.4.3 backports_1.5.0 R6_2.6.1 lazyeval_0.2.2 uwot_0.2.4 withr_3.0.2 gridExtra_2.3 progressr_0.18.0 cli_3.6.5 textshaping_1.0.4 spatstat.explore_3.7-0 fastDummies_1.7.5 scatterpie_0.2.6
## [22] labeling_0.4.3 sass_0.4.10 S7_0.2.1 spatstat.data_3.1-9 ggridges_0.5.7 pbapply_1.7-4 pkgdown_2.2.0 yulab.utils_0.2.4 Rsamtools_2.26.0 systemfonts_1.3.1 gson_0.1.0 foreign_0.8-91 R.utils_2.13.0 DOSE_4.4.0 dichromat_2.0-0.1 parallelly_1.46.1 limma_3.66.0 BSgenome_1.78.0 rstudioapi_0.18.0 RSQLite_2.4.6 gridGraphics_0.5-1
## [43] BiocIO_1.20.0 ica_1.0-3 spatstat.random_3.4-4 dplyr_1.2.0 GO.db_3.22.0 Matrix_1.7-4 ggbeeswarm_0.7.3 abind_1.4-8 R.methodsS3_1.8.2 lifecycle_1.0.5 yaml_2.3.12 SummarizedExperiment_1.40.0 qvalue_2.42.0 SparseArray_1.10.8 Rtsne_0.17 grid_4.5.1 blob_1.3.0 promises_1.5.0 crayon_1.5.3 ggtangle_0.1.1 miniUI_0.1.2
## [64] lattice_0.22-9 cowplot_1.2.0 cigarillo_1.0.0 KEGGREST_1.50.0 pillar_1.11.1 knitr_1.51 fgsea_1.36.2 rjson_0.2.23 future.apply_1.20.2 codetools_0.2-20 fastmatch_1.1-8 glue_1.8.0 ggiraph_0.9.6 fontLiberation_0.1.0 ggfun_0.2.0 spatstat.univar_3.1-6 data.table_1.18.2.1 treeio_1.34.0 vctrs_0.7.2 png_0.1-9 spam_2.11-3
## [85] gtable_0.3.6 cachem_1.1.0 xfun_0.57 S4Arrays_1.10.1 mime_0.13 survival_3.8-6 RcppRoll_0.3.2 statmod_1.5.1 fitdistrplus_1.2-6 ROCR_1.0-12 nlme_3.1-168 ggtree_4.0.4 fontquiver_0.2.1 bit64_4.6.0-1 filelock_1.0.3 RcppAnnoy_0.0.23 bslib_0.10.0 irlba_2.3.7 vipor_0.4.7 KernSmooth_2.23-26 otel_0.2.0
## [106] rpart_4.1.24 colorspace_2.1-2 DBI_1.3.0 Hmisc_5.2-5 nnet_7.3-20 ggrastr_1.0.2 tidyselect_1.2.1 bit_4.6.0 compiler_4.5.1 curl_7.0.0 httr2_1.2.2 htmlTable_2.4.3 hdf5r_1.3.12 fontBitstreamVera_0.1.1 desc_1.4.3 DelayedArray_0.36.0 plotly_4.12.0 stringfish_0.18.0 rtracklayer_1.70.1 checkmate_2.3.4 scales_1.4.0
## [127] lmtest_0.9-40 rappdirs_0.3.4 stringr_1.6.0 digest_0.6.39 goftest_1.2-3 presto_1.0.0 spatstat.utils_3.2-1 rmarkdown_2.31 XVector_0.50.0 htmltools_0.5.9 pkgconfig_2.0.3 base64enc_0.1-6 sparseMatrixStats_1.22.0 MatrixGenerics_1.22.0 fastmap_1.2.0 rlang_1.1.7 htmlwidgets_1.6.4 UCSC.utils_1.6.1 shiny_1.13.0 farver_2.1.2 jquerylib_0.1.4
## [148] zoo_1.8-15 jsonlite_2.0.0 BiocParallel_1.44.0 R.oo_1.27.1 GOSemSim_2.36.0 VariantAnnotation_1.56.0 RCurl_1.98-1.18 magrittr_2.0.4 ggplotify_0.1.3 Formula_1.2-5 dotCall64_1.2 Rcpp_1.1.1 gdtools_0.5.0 ape_5.8-1 ggnewscale_0.5.2 reticulate_1.45.0 stringi_1.8.7 MASS_7.3-65 plyr_1.8.9 parallel_4.5.1 listenv_0.10.1
## [169] ggrepel_0.9.7 deldir_2.0-4 Biostrings_2.78.0 splines_4.5.1 tensor_1.5.1 igraph_2.2.2 spatstat.geom_3.7-0 RcppHNSW_0.6.0 reshape2_1.4.5 qs2_0.1.7 BiocVersion_3.22.0 XML_3.99-0.23 evaluate_1.0.5 biovizBase_1.58.0 RcppParallel_5.1.11-1 BiocManager_1.30.27 tweenr_2.0.3 httpuv_1.6.16 RANN_2.6.2 tidyr_1.3.2 purrr_1.2.1
## [190] polyclip_1.10-7 scattermore_1.2 ggforce_0.5.0 xtable_1.8-8 restfulr_0.0.16 tidytree_0.4.7 RSpectra_0.16-2 tidydr_0.0.6 later_1.4.7 viridisLite_0.4.3 ragg_1.5.0 tibble_3.3.1 aplot_0.2.9 beeswarm_0.4.0 memoise_2.0.1 GenomicAlignments_1.46.0 cluster_2.1.8.2 globals_0.19.1