Motif analysis with Signac
Compiled: April 01, 2026
Source:vignettes/motif_vignette.Rmd
motif_vignette.RmdIn this tutorial, we will perform DNA sequence motif analysis in Signac. We will explore two complementary options for performing motif analysis: one by finding overrepresented motifs in a set of differentially accessible peaks, one method performing differential motif activity analysis between groups of cells.
In this demonstration we use data from the adult mouse brain. See our vignette for the code used to generate this object, and links to the raw data. First, load the required packages and the pre-computed Seurat object:
library(Signac)
library(Seurat)
library(JASPAR2020)
library(TFBSTools)
library(BSgenome.Mmusculus.UCSC.mm10)
library(patchwork)
mouse_brain <- qs2::qs_read("adult_mouse_brain.qs2")
mouse_brain## An object of class Seurat
## 179011 features across 3512 samples within 2 assays
## Active assay: peaks (157203 features, 157203 variable features)
## 2 layers present: counts, data
## 1 other assay present: RNA
## 2 dimensional reductions calculated: lsi, umap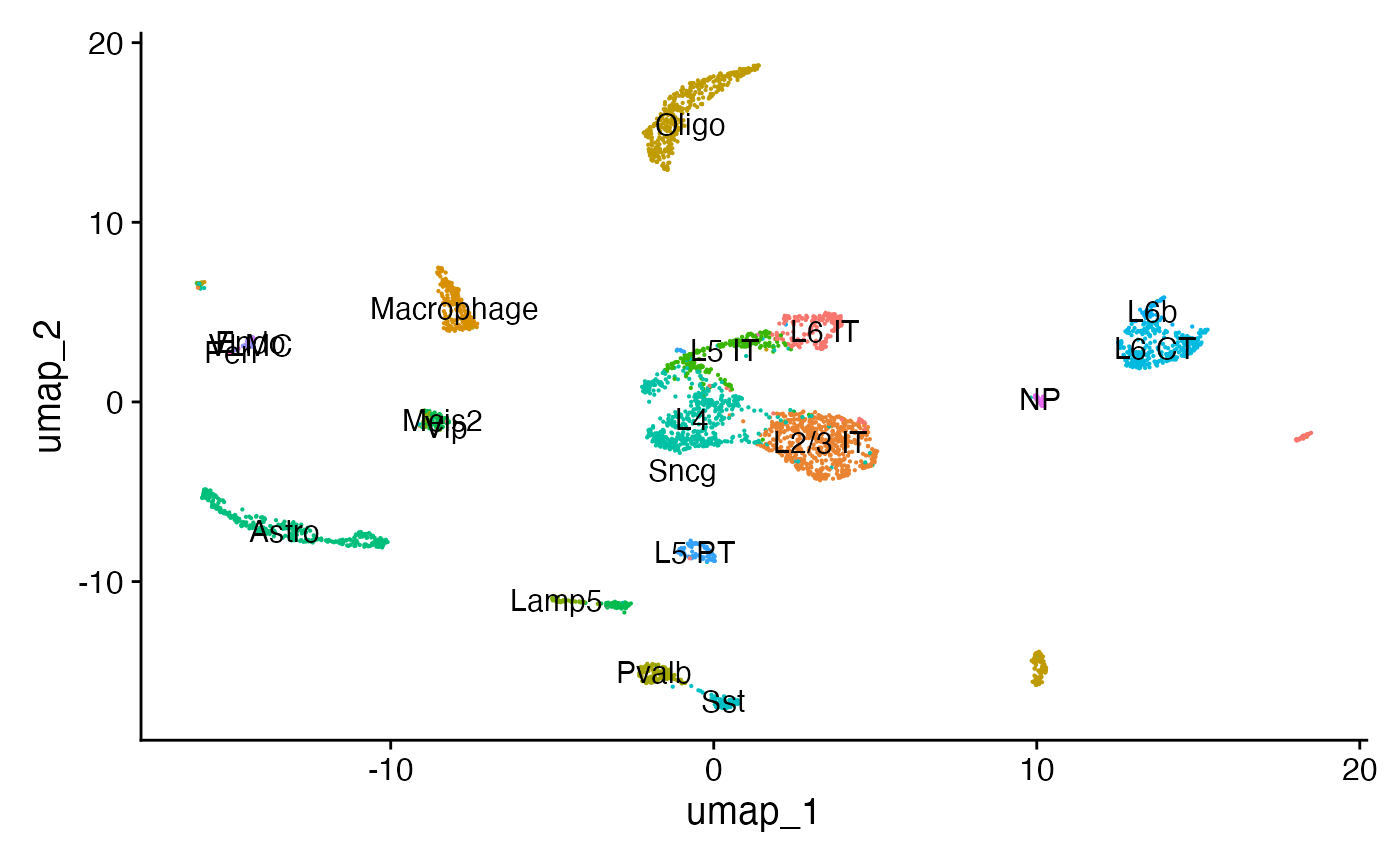
Adding motif information to the Seurat object
To add the DNA sequence motif information required for motif
analyses, we can run the AddMotifs() function:
# Get a list of motif position frequency matrices from the JASPAR database
pfm <- getMatrixSet(
x = JASPAR2020,
opts = list(collection = "CORE", tax_group = 'vertebrates', all_versions = FALSE)
)
# add motif information
mouse_brain <- AddMotifs(
object = mouse_brain,
genome = BSgenome.Mmusculus.UCSC.mm10,
pfm = pfm
)To facilitate motif analysis in Signac, we have created the
Motif class to store all the required information,
including a list of position weight matrices (PWMs) or position
frequency matrices (PFMs) and a motif occurrence matrix. Here, the
AddMotifs() function constructs a Motif object
and adds it to our mouse brain dataset, along with other information
such as the base composition of each peak. A motif object can be added
to any Seurat assay using the SetAssayData() function. See
the object interaction vignette for
more information.
Finding overrepresented motifs
To identify potentially important cell-type-specific regulatory sequences, we can search for DNA motifs that are overrepresented in a set of peaks that are differentially accessible between cell types.
Here, we find differentially accessible peaks between Pvalb and Sst
inhibitory interneurons. For sparse data (such as scATAC-seq), we find
it is often necessary to lower the min.pct threshold in
FindMarkers() from the default (0.1, which was designed for
scRNA-seq data).
We then perform a hypergeometric test to test the probability of observing the motif at the given frequency by chance, comparing with a background set of peaks matched for GC content.
da_peaks <- FindMarkers(
object = mouse_brain,
ident.1 = 'Pvalb',
ident.2 = 'Sst',
only.pos = TRUE,
test.use = 'LR',
min.pct = 0.05,
latent.vars = 'nCount_peaks'
)
# get top differentially accessible peaks
top.da.peak <- rownames(da_peaks[da_peaks$p_val < 0.005 & da_peaks$pct.1 > 0.2, ])Optional: choosing a set of background peaks
Matching the set of background peaks is essential when finding enriched DNA sequence motifs. By default, we choose a set of peaks matched for GC content, but it can be sometimes be beneficial to further restrict the background peaks to those that are accessible in the groups of cells compared when finding differentially accessible peaks.
The AccessiblePeaks() function can be used to find a set
of peaks that are open in a subset of cells. We can use this function to
first restrict the set of possible background peaks to those peaks that
were open in the set of cells compared in FindMarkers(),
and then create a GC-content-matched set of peaks from this larger set
using MatchRegionStats().
# find peaks open in Pvalb or Sst cells
open.peaks <- AccessiblePeaks(mouse_brain, idents = c("Pvalb", "Sst"))
# match the overall GC content in the peak set
meta.feature <- GetAssayData(mouse_brain, assay = "peaks", layer = "meta.features")
peaks.matched <- MatchRegionStats(
meta.feature = meta.feature[open.peaks, ],
query.feature = meta.feature[top.da.peak, ],
n = 50000
)## Matching GC.percent distributionpeaks.matched can then be used as the background peak
set by setting background=peaks.matched in
FindMotifs().
# test enrichment
enriched.motifs <- FindMotifs(
object = mouse_brain,
features = top.da.peak
)## Selecting background regions to match input sequence characteristics## Matching GC.percent distribution## Testing motif enrichment in 1131 regions| motif | observed | background | percent.observed | percent.background | fold.enrichment | pvalue | motif.name | p.adjust | |
|---|---|---|---|---|---|---|---|---|---|
| MA0497.1 | MA0497.1 | 556 | 8315 | 49.16004 | 20.7875 | 2.364884 | 0 | MEF2C | 0 |
| MA0052.4 | MA0052.4 | 533 | 7948 | 47.12644 | 19.8700 | 2.371738 | 0 | MEF2A | 0 |
| MA0773.1 | MA0773.1 | 398 | 4930 | 35.19010 | 12.3250 | 2.855180 | 0 | MEF2D | 0 |
| MA0660.1 | MA0660.1 | 345 | 4002 | 30.50398 | 10.0050 | 3.048873 | 0 | MEF2B | 0 |
| MA1151.1 | MA1151.1 | 286 | 3206 | 25.28736 | 8.0150 | 3.155004 | 0 | RORC | 0 |
| MA0592.3 | MA0592.3 | 350 | 4669 | 30.94607 | 11.6725 | 2.651194 | 0 | ESRRA | 0 |
We can also plot the position weight matrices for the motifs, so we can visualize the different motif sequences.
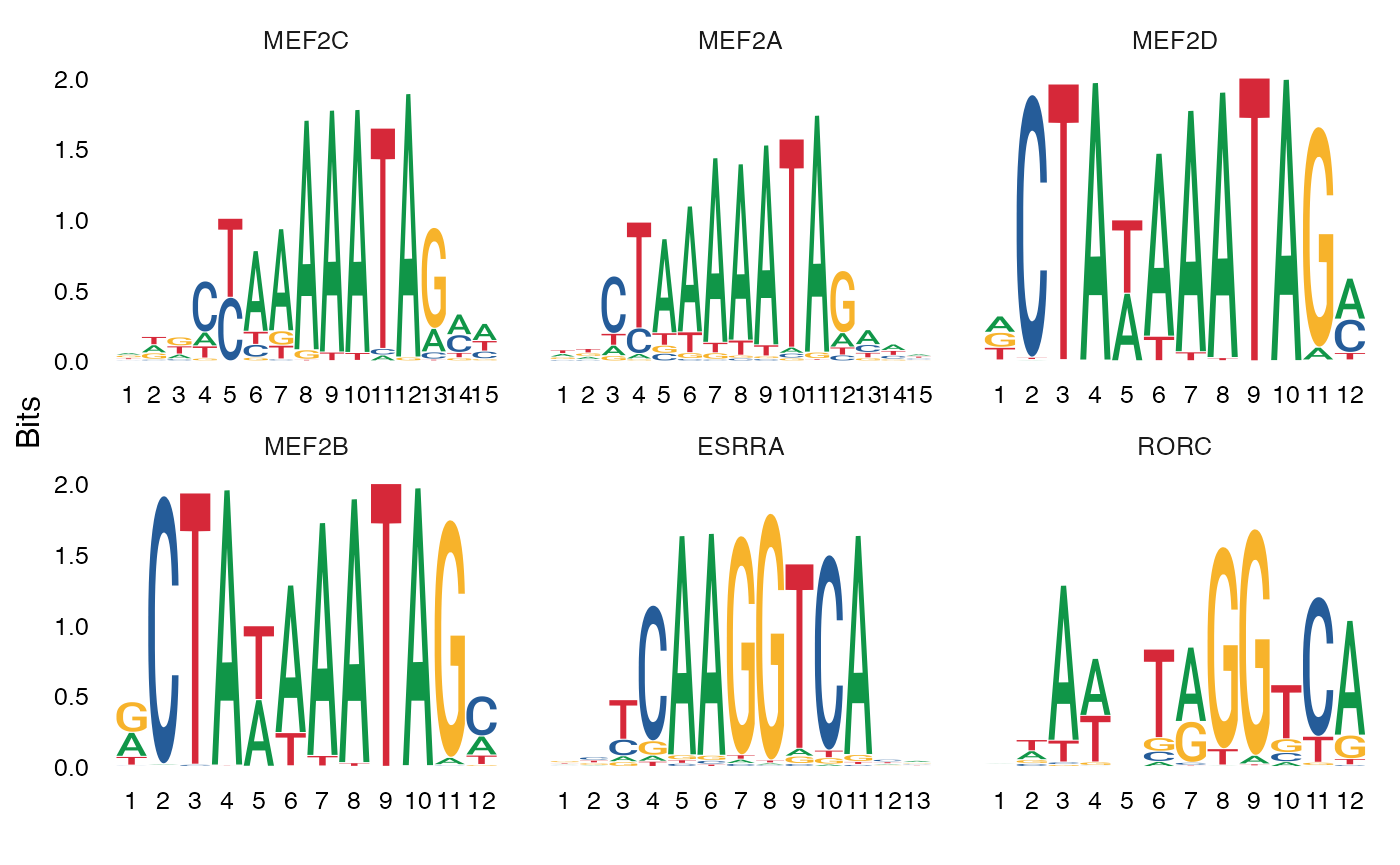
Mef-family motifs, particularly Mef2c, are enriched in
Pvalb-specific peaks in scATAC-seq data (https://doi.org/10.1016/j.cell.2019.05.031; https://doi.org/10.1101/615179), and Mef2c is
required for the development of Pvalb interneurons (https://www.nature.com/articles/nature25999). Here our
results are consistent with these findings, and we observe a strong
enrichment of Mef-family motifs in the top results from
FindMotifs().
Session Info
## R version 4.5.1 (2025-06-13)
## Platform: aarch64-apple-darwin20
## Running under: macOS Tahoe 26.4
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: Asia/Singapore
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] patchwork_1.3.2 BSgenome.Mmusculus.UCSC.mm10_1.4.3
## [3] BSgenome_1.78.0 rtracklayer_1.70.1
## [5] BiocIO_1.20.0 Biostrings_2.78.0
## [7] XVector_0.50.0 GenomicRanges_1.62.1
## [9] Seqinfo_1.0.0 IRanges_2.44.0
## [11] S4Vectors_0.48.0 BiocGenerics_0.56.0
## [13] generics_0.1.4 TFBSTools_1.48.0
## [15] JASPAR2020_0.99.10 Seurat_5.4.0
## [17] SeuratObject_5.3.0.9002 sp_2.2-1
## [19] Signac_1.17.0
##
## loaded via a namespace (and not attached):
## [1] RcppAnnoy_0.0.23 splines_4.5.1
## [3] later_1.4.7 bitops_1.0-9
## [5] tibble_3.3.1 polyclip_1.10-7
## [7] XML_3.99-0.23 DirichletMultinomial_1.52.0
## [9] fastDummies_1.7.5 lifecycle_1.0.5
## [11] pwalign_1.6.0 globals_0.19.1
## [13] lattice_0.22-9 MASS_7.3-65
## [15] magrittr_2.0.4 plotly_4.12.0
## [17] sass_0.4.10 rmarkdown_2.31
## [19] jquerylib_0.1.4 yaml_2.3.12
## [21] httpuv_1.6.16 otel_0.2.0
## [23] sctransform_0.4.3 spam_2.11-3
## [25] spatstat.sparse_3.1-0 reticulate_1.45.0
## [27] cowplot_1.2.0 pbapply_1.7-4
## [29] DBI_1.3.0 RColorBrewer_1.1-3
## [31] abind_1.4-8 Rtsne_0.17
## [33] purrr_1.2.1 RCurl_1.98-1.18
## [35] ggrepel_0.9.7 irlba_2.3.7
## [37] listenv_0.10.1 spatstat.utils_3.2-1
## [39] seqLogo_1.76.0 goftest_1.2-3
## [41] RSpectra_0.16-2 spatstat.random_3.4-4
## [43] fitdistrplus_1.2-6 parallelly_1.46.1
## [45] pkgdown_2.2.0 codetools_0.2-20
## [47] DelayedArray_0.36.0 RcppRoll_0.3.2
## [49] wrswoR_1.2.0 tidyselect_1.2.1
## [51] ggseqlogo_0.2.2 UCSC.utils_1.6.1
## [53] farver_2.1.2 matrixStats_1.5.0
## [55] spatstat.explore_3.7-0 GenomicAlignments_1.46.0
## [57] jsonlite_2.0.0 progressr_0.18.0
## [59] motifmatchr_1.32.0 ggridges_0.5.7
## [61] survival_3.8-6 systemfonts_1.3.1
## [63] tools_4.5.1 ragg_1.5.0
## [65] TFMPvalue_1.0.0 ica_1.0-3
## [67] Rcpp_1.1.1 glue_1.8.0
## [69] gridExtra_2.3 SparseArray_1.10.8
## [71] qs2_0.1.7 xfun_0.57
## [73] MatrixGenerics_1.22.0 GenomeInfoDb_1.46.2
## [75] dplyr_1.2.0 withr_3.0.2
## [77] fastmap_1.2.0 caTools_1.18.3
## [79] digest_0.6.39 R6_2.6.1
## [81] mime_0.13 textshaping_1.0.4
## [83] scattermore_1.2 tensor_1.5.1
## [85] gtools_3.9.5 spatstat.data_3.1-9
## [87] dichromat_2.0-0.1 RSQLite_2.4.6
## [89] cigarillo_1.0.0 tidyr_1.3.2
## [91] data.table_1.18.2.1 httr_1.4.8
## [93] htmlwidgets_1.6.4 S4Arrays_1.10.1
## [95] uwot_0.2.4 pkgconfig_2.0.3
## [97] gtable_0.3.6 blob_1.3.0
## [99] lmtest_0.9-40 S7_0.2.1
## [101] htmltools_0.5.9 dotCall64_1.2
## [103] logging_0.10-108 scales_1.4.0
## [105] Biobase_2.70.0 png_0.1-9
## [107] spatstat.univar_3.1-6 knitr_1.51
## [109] reshape2_1.4.5 rjson_0.2.23
## [111] nlme_3.1-168 curl_7.0.0
## [113] cachem_1.1.0 zoo_1.8-15
## [115] stringr_1.6.0 KernSmooth_2.23-26
## [117] parallel_4.5.1 miniUI_0.1.2
## [119] restfulr_0.0.16 desc_1.4.3
## [121] pillar_1.11.1 grid_4.5.1
## [123] vctrs_0.7.2 RANN_2.6.2
## [125] promises_1.5.0 stringfish_0.18.0
## [127] xtable_1.8-8 cluster_2.1.8.2
## [129] evaluate_1.0.5 cli_3.6.5
## [131] compiler_4.5.1 Rsamtools_2.26.0
## [133] rlang_1.1.7 crayon_1.5.3
## [135] future.apply_1.20.2 labeling_0.4.3
## [137] plyr_1.8.9 fs_2.0.1
## [139] stringi_1.8.7 deldir_2.0-4
## [141] viridisLite_0.4.3 BiocParallel_1.44.0
## [143] lazyeval_0.2.2 spatstat.geom_3.7-0
## [145] Matrix_1.7-4 RcppHNSW_0.6.0
## [147] sparseMatrixStats_1.22.0 bit64_4.6.0-1
## [149] future_1.70.0 ggplot2_4.0.2
## [151] shiny_1.13.0 SummarizedExperiment_1.40.0
## [153] ROCR_1.0-12 igraph_2.2.2
## [155] memoise_2.0.1 RcppParallel_5.1.11-1
## [157] bslib_0.10.0 fastmatch_1.1-8
## [159] bit_4.6.0