Every cell within an organism carries essentially the same genetic information. In multicellular organisms, this common genetic sequence is decoded differently by different cell types to produce a multitude of cell states, each performing specialized and essential functions. How these different cell states are encoded in the genome is a major question yet to be fully understood. However, we do know that chromatin states are highly dynamic and instrumental in guiding the proper development of these cellular phenotypic states. Our lab aims to develop novel approaches to study how chromatin states guide cell fates, how these healthy developmental processes are impacted by natural human genetic variation, and how pathogenic somatic mutations perturb healthy cell development.
Molecular technologies
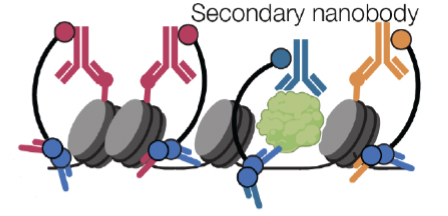
We develop new molecular technologies to study how the genome is packaged and decoded within cells. In 2022, we introduced nanobody-tethered transposition sequencing (NTT-seq), a transposition-based chromatin profiling method that enables the simultaneous measurement of multiple histone posttranslation modifications genome-wide with single-cell resolution. Check out the NTT-seq website for more. We are working on further improvements to increase the scalability, cell throughput, and sensitivity of NTT-seq, as well as developing new molecular assays to profile chromatin states at single-cell resolution. Furthermore, we are developing new methods of perturbing cellular chromatin states to advance our understanding of the function of different aspects of chromatin state, including the presence of different histone posttranslational modifications and three-dimensional chromatin structure.
Computational methods for single-cell genomics
We develop new computational methods for genomics with a focus on methods for the processing and analysis of single-cell epigenomic datasets. We currently develop and maintain the Signac and Sinto packages for the processing and analysis of single-cell genomic data. Signac is a comprehensive R package that supports quality control, peak calling, quantification, dimension reduction, differential testing, specialized data visualization, motif analysis, and data integration tasks for single-cell epigenomics. Sinto is a Python package providing utilities commonly needed for single-cell analysis and the processing of raw single-cell sequencing data. We continue to work on new computational methods for single-cell epigenomics to complement the new experimental methods that are available, such as NTT-seq, and incorporate these methods into open-source software packages.
Visit the Stuart lab GitHub page for more.
Epigenetics of human blood cancers
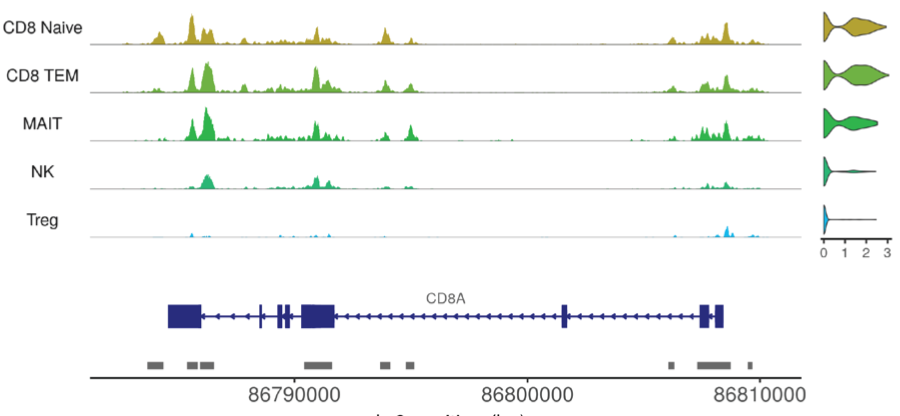
Aberrant epigenomic regulation has been strongly implicated in several cancers, including leukemia, lymphoma, and myeloma. Mutations in key chromatin regulators such as EZH2, MLL genes, DNMT3A, TET2, and ASXL1 are all associated with the development of blood cancers affecting millions of people worldwide. However, it remains unclear how the function of these chromatin regulator proteins are affected by DNA mutations, and how chromatin states are impacted by altered regulator function during cell development. Through collaborations with clinicians at the Cancer Science Institute of Singapore and Duke-NUS medical school we are applying single-cell chromatin profiling methods, including scNTT-seq and scATAC-seq, to study how mutations in epigenomic regulators impact the state of human blood cells in leukemia, lymphoma, and myeloma.
Funding
Current support
| Date | Project title | Funding organization | Scheme | Project number |
|---|---|---|---|---|
| 2025 | Saturation mutagenesis to study Asian variants of unknown significance (VUS) | A*STAR | Council Strategic Fund | C250314064 |
| 2024 - 2029 | Design of synthetic DNA regulatory elements for precision gene therapy | NRF | Fellowship | NRF-NRFF16-2024-0005 |
Past support
| Date | Project title | Funding organization | Scheme | Project number |
|---|---|---|---|---|
| 2021 - 2023 | Comprehensive mapping of multimodal chromatin state in single cells | NIH - NHGRI | K99/R00 | 1K99HG011489 |