scATAC-seq data integration
Compiled: April 01, 2026
Source:vignettes/integrate_atac.Rmd
integrate_atac.RmdHere we demonstrate the integration of multiple single-cell chromatin datasets derived from human PBMCs. One dataset was generated using the 10x Genomics multiome technology, and includes DNA accessibility and gene expression information for each cell. The other dataset was profiled using 10x Genomics scATAC-seq, and includes DNA accessibility data only.
We will integrate the two datasets together using the shared DNA accessibility assay, using tools available in the Seurat package. Furthermore, we will demonstrate transferring both continuous (gene expression) and categorical (cell labels) information from a reference to a query single-cell chromatin dataset.
View data download code
The PBMC multiome and scATAC-seq data can be downloaded from the 10x website:
# multiome
wget https://cf.10xgenomics.com/samples/cell-arc/1.0.0/pbmc_granulocyte_sorted_10k/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5
wget https://cf.10xgenomics.com/samples/cell-arc/1.0.0/pbmc_granulocyte_sorted_10k/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz
wget https://cf.10xgenomics.com/samples/cell-arc/1.0.0/pbmc_granulocyte_sorted_10k/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz.tbi
# scATAC
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_filtered_peak_bc_matrix.h5
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_singlecell.csv
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz
wget https://cf.10xgenomics.com/samples/cell-atac/2.1.0/10k_pbmc_ATACv2_nextgem_Chromium_Controller/10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz.tbiPreprocessing
Here we’ll load the PBMC multiome data pre-processed in our multiome vignette and the PBMC scATAC-seq data from our PBMC vignette:
library(Signac)
library(Seurat)
library(ggplot2)
# load the pre-processed multiome data
pbmc.multi <- qs2::qs_read("multiomic/pbmc_multiomic.qs2")
# load the pre-processed atac data
pbmc.atac <- qs2::qs_read("pbmc_vignette/pbmc.qs2")An important first step in any integrative analysis of single-cell chromatin data is to ensure that the same features are measured in each dataset. Here, we quantify the multiome peaks in the ATAC dataset to ensure that there are common features across the two datasets. See the merge vignette for more information about merging chromatin assays.
# quantify multiome peaks in the scATAC-seq dataset
counts <- FeatureMatrix(
fragments = Fragments(pbmc.atac),
features = granges(pbmc.multi),
cells = colnames(pbmc.atac)
)
# add new assay with multiome peaks
pbmc.atac[['ATAC']] <- CreateChromatinAssay(
counts = counts,
fragments = Fragments(pbmc.atac)
)
# compute LSI
DefaultAssay(pbmc.atac) <- "ATAC"
pbmc.atac <- FindTopFeatures(pbmc.atac, min.cutoff = 10)
pbmc.atac <- RunTFIDF(pbmc.atac)
pbmc.atac <- RunSVD(pbmc.atac)Next we can merge the multiome and scATAC datasets together and observe that there is a difference between them that appears to be due to the batch (experiment and technology-specific variation).
# first add dataset-identifying metadata
pbmc.atac$dataset <- "ATAC"
pbmc.multi$dataset <- "Multiome"
# merge
pbmc.combined <- merge(pbmc.atac, pbmc.multi)
# process the combined dataset
pbmc.combined <- FindTopFeatures(pbmc.combined, min.cutoff = 10)
pbmc.combined <- RunTFIDF(pbmc.combined)
pbmc.combined <- RunSVD(pbmc.combined)
pbmc.combined <- RunUMAP(pbmc.combined, reduction = "lsi", dims = 2:30)
p1 <- DimPlot(pbmc.combined, group.by = "dataset")Integration
To find integration anchors between the two datasets, we need to
project them into a shared low-dimensional space. To do this, we’ll use
reciprocal LSI projection (projecting each dataset into the others LSI
space) by setting reduction="rlsi". For more information
about the data integration methods in Seurat, see our recent paper and the Seurat website.
Rather than integrating the normalized data matrix, as is typically
done for scRNA-seq data, we’ll integrate the low-dimensional cell
embeddings (the LSI coordinates) across the datasets using the
IntegrateEmbeddings() function. This is much better suited
to scATAC-seq data, as we typically have a very sparse matrix with a
large number of features. Note that this requires that we first compute
an uncorrected LSI embedding using the merged dataset (as we did
above).
# find integration anchors
integration.anchors <- FindIntegrationAnchors(
object.list = list(pbmc.multi, pbmc.atac),
anchor.features = rownames(pbmc.multi),
reduction = "rlsi",
dims = 2:30
)
# integrate LSI embeddings
integrated <- IntegrateEmbeddings(
anchorset = integration.anchors,
reductions = pbmc.combined[["lsi"]],
new.reduction.name = "integrated_lsi",
dims.to.integrate = 1:30
)
# create a new UMAP using the integrated embeddings
integrated <- RunUMAP(integrated, reduction = "integrated_lsi", dims = 2:30)
p2 <- DimPlot(integrated, group.by = "dataset")Finally, we can compare the results of the merged and integrated datasets, and find that the integration has successfully removed the technology-specific variation in the dataset while retaining the cell-type-specific (biological) variation.
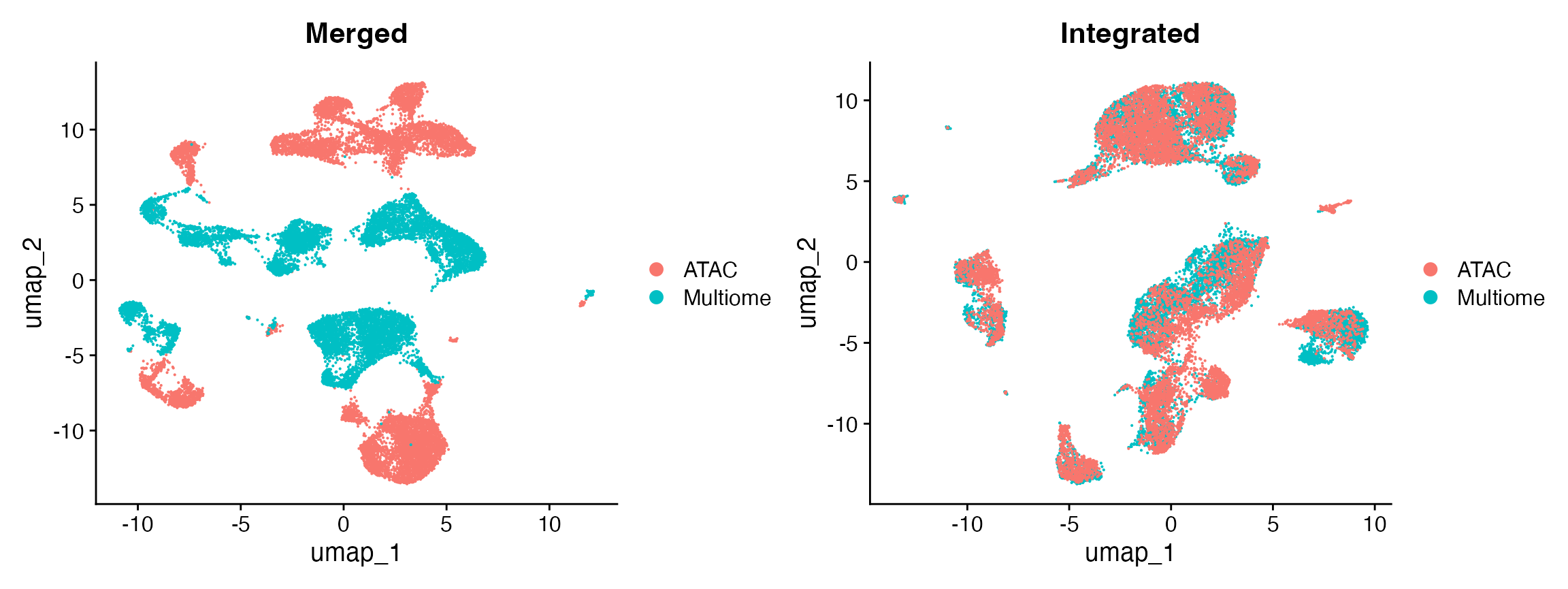
Here we’ve demonstrated the integration method using two datasets, but the same workflow can be applied to integrate any number of datasets.
Reference mapping
In cases where we have a large, high-quality dataset, or a dataset containing unique information not present in other datasets (cell type annotations or additional data modalities, for example), we often want to use that dataset as a reference and map queries onto it so that we can interpret these query datasets in the context of the existing reference.
To demonstrate how to do this using single-cell chromatin reference
and query datasets, we’ll treat the PBMC multiome dataset here as a
reference and map the scATAC-seq dataset to it using the
FindTransferAnchors() and MapQuery() functions
from Seurat.
# compute UMAP and store the UMAP model
pbmc.multi <- RunUMAP(pbmc.multi, reduction = "lsi", dims = 2:30, return.model = TRUE)
# find transfer anchors
transfer.anchors <- FindTransferAnchors(
reference = pbmc.multi,
query = pbmc.atac,
reference.reduction = "lsi",
reduction = "lsiproject",
dims = 2:30
)
# map query onto the reference dataset
pbmc.atac <- MapQuery(
anchorset = transfer.anchors,
reference = pbmc.multi,
query = pbmc.atac,
refdata = pbmc.multi$predicted.id,
reference.reduction = "lsi",
new.reduction.name = "ref.lsi",
reduction.model = 'umap'
)What is MapQuery() doing?
MapQuery() is a wrapper function that runs
TransferData(), IntegrateEmbeddings(), and
ProjectUMAP() for a query dataset, and sets sensible
default parameters based on how the anchor object was generated. For
finer control over the parameters used by each of these functions, you
can pass parameters through MapQuery() to each function
using the transferdata.args,
integrateembeddings.args, and projectumap.args
arguments for MapQuery(), or you can run each of the
functions yourself. For example:
pbmc.atac <- TransferData(
anchorset = transfer.anchors,
reference = pbmc.multi,
weight.reduction = "lsiproject",
query = pbmc.atac,
refdata = list(
celltype = "predicted.id",
predicted_RNA = "RNA")
)
pbmc.atac <- IntegrateEmbeddings(
anchorset = transfer.anchors,
reference = pbmc.multi,
query = pbmc.atac,
reductions = "lsiproject",
new.reduction.name = "ref.lsi"
)
pbmc.atac <- ProjectUMAP(
query = pbmc.atac,
query.reduction = "ref.lsi",
reference = pbmc.multi,
reference.reduction = "lsi",
reduction.model = "umap"
)By running MapQuery(), we have mapped the scATAC-seq
dataset onto the the multimodal reference, and enabled cell type labels
to be transferred from reference to query. We can visualize these
reference mapping results and the cell type labels now associated with
the scATAC-seq dataset:
p1 <- DimPlot(pbmc.multi, reduction = "umap", group.by = "predicted.id", label = TRUE, repel = TRUE) + NoLegend() + ggtitle("Reference")
p2 <- DimPlot(pbmc.atac, reduction = "ref.umap", group.by = "predicted.id", label = TRUE, repel = TRUE) + NoLegend() + ggtitle("Query")
p1 | p2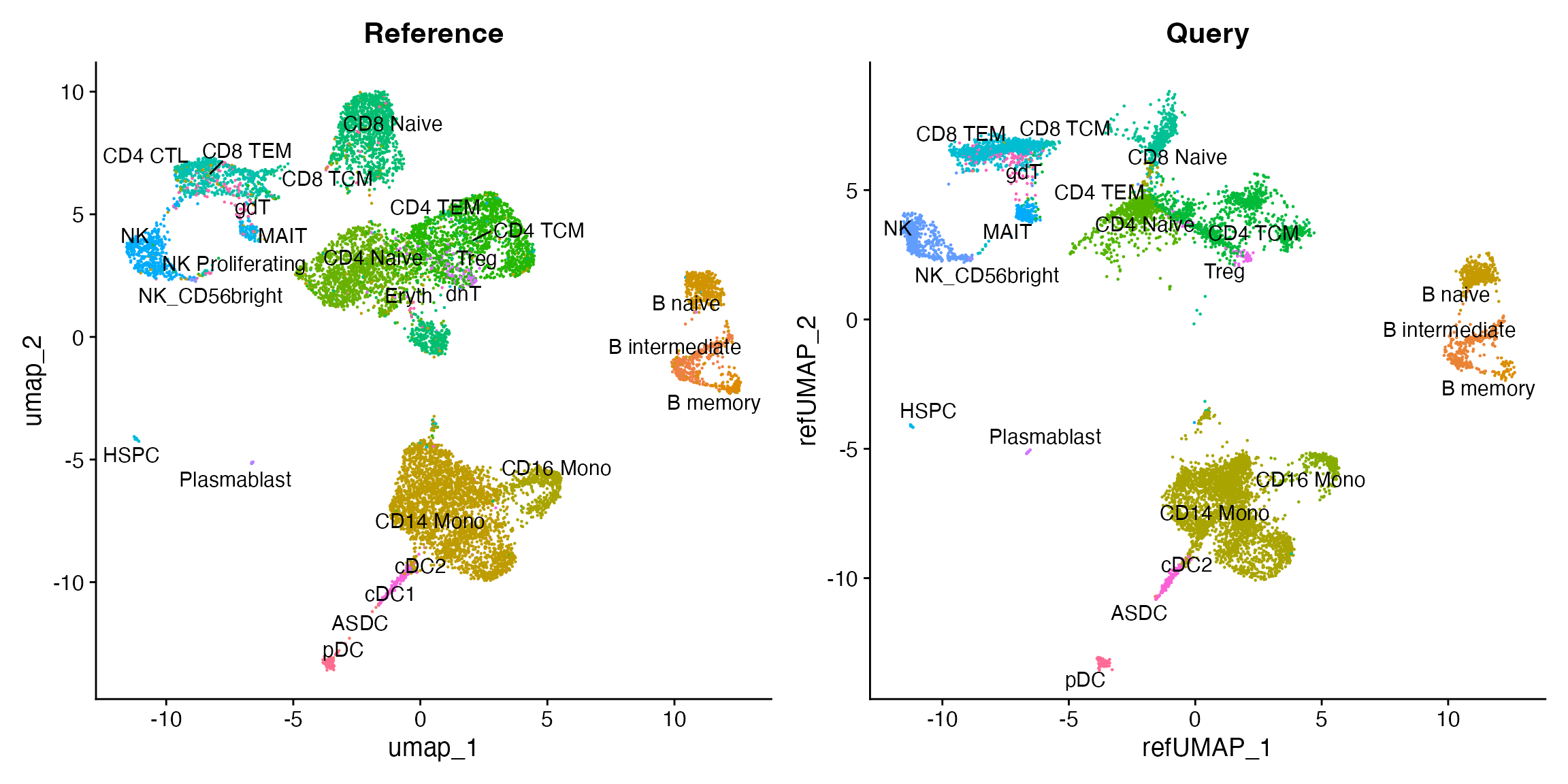
For more information about multimodal reference mapping, see the Seurat vignette.
RNA imputation
Above we transferred categorical information (the cell labels) and
mapped the query data onto an existing reference UMAP. We can also
transfer continuous data from the reference to the query in the same
way. Here we demonstrate transferring the gene expression values from
the PBMC multiome dataset (that measured DNA accessibility and gene
expression in the same cells) to the PBMC scATAC-seq dataset (that
measured DNA accessibility only). Note that we could also transfer these
values using the MapQuery() function call above by setting
the refdata parameter to a list of values.
# predict gene expression values
rna <- TransferData(
anchorset = transfer.anchors,
refdata = LayerData(pbmc.multi, assay = "SCT", layer = "data"),
weight.reduction = pbmc.atac[["lsi"]],
dims = 2:30
)
# add predicted values as a new assay
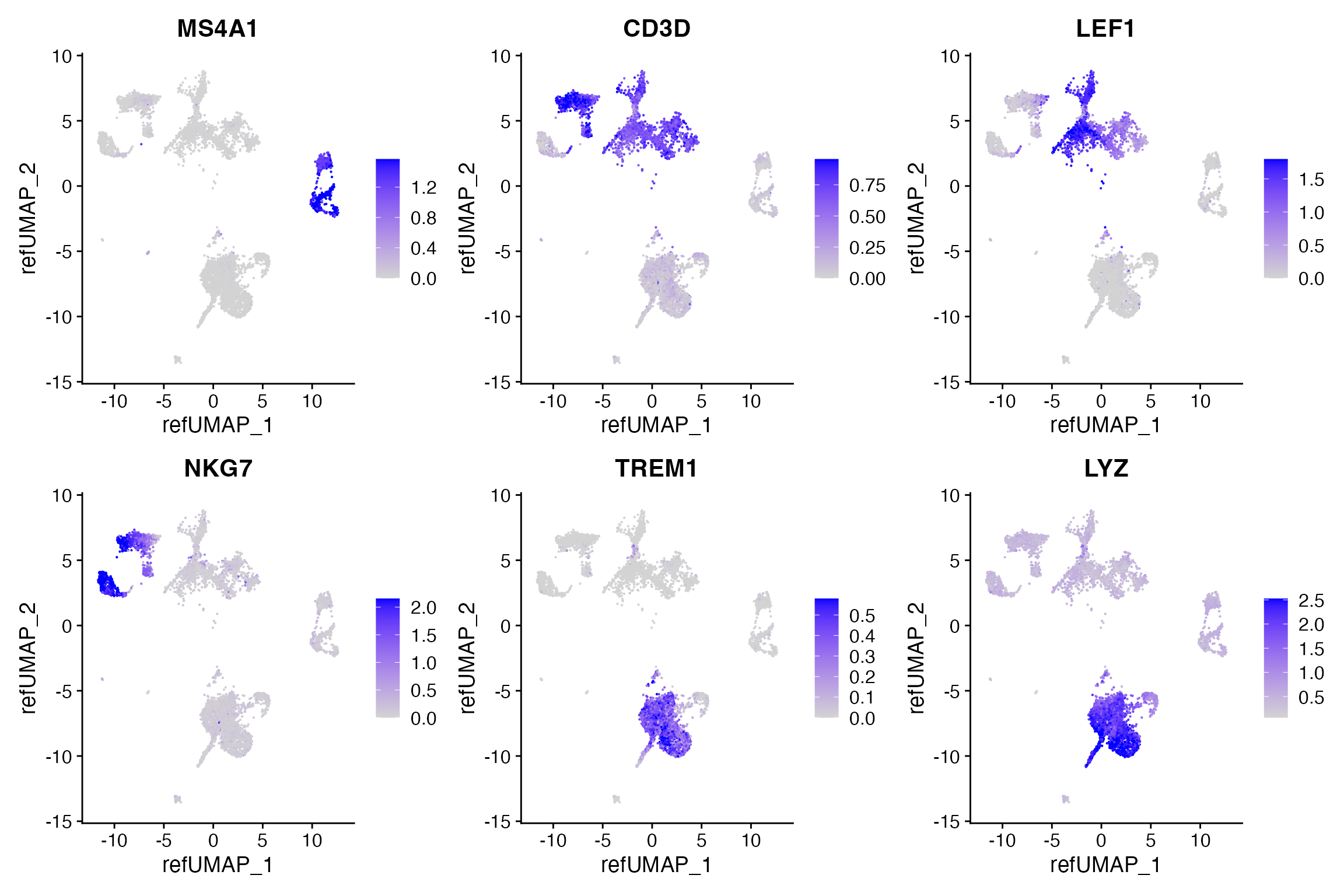
pbmc.atac[["predicted"]] <- rnaWe can look at some immune marker genes and see that the predicted expression patterns match our expectation based on known expression patterns.
DefaultAssay(pbmc.atac) <- "predicted"
FeaturePlot(
object = pbmc.atac,
features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),
pt.size = 0.1,
max.cutoff = 'q95',
reduction = "ref.umap",
ncol = 3
)
Session Info
## R version 4.5.1 (2025-06-13)
## Platform: aarch64-apple-darwin20
## Running under: macOS Tahoe 26.4
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: Asia/Singapore
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] future_1.70.0 ggplot2_4.0.2 Seurat_5.4.0
## [4] SeuratObject_5.3.0.9002 sp_2.2-1 Signac_1.17.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.4
## [4] spatstat.utils_3.2-1 farver_2.1.2 rmarkdown_2.31
## [7] fs_2.0.1 ragg_1.5.0 vctrs_0.7.2
## [10] ROCR_1.0-12 spatstat.explore_3.7-0 Rsamtools_2.26.0
## [13] RcppRoll_0.3.2 htmltools_0.5.9 sass_0.4.10
## [16] sctransform_0.4.3 parallelly_1.46.1 KernSmooth_2.23-26
## [19] bslib_0.10.0 htmlwidgets_1.6.4 desc_1.4.3
## [22] ica_1.0-3 plyr_1.8.9 plotly_4.12.0
## [25] zoo_1.8-15 cachem_1.1.0 igraph_2.2.2
## [28] mime_0.13 lifecycle_1.0.5 pkgconfig_2.0.3
## [31] Matrix_1.7-4 R6_2.6.1 fastmap_1.2.0
## [34] MatrixGenerics_1.22.0 fitdistrplus_1.2-6 shiny_1.13.0
## [37] digest_0.6.39 patchwork_1.3.2 S4Vectors_0.48.0
## [40] tensor_1.5.1 RSpectra_0.16-2 irlba_2.3.7
## [43] textshaping_1.0.4 GenomicRanges_1.62.1 labeling_0.4.3
## [46] progressr_0.18.0 spatstat.sparse_3.1-0 polyclip_1.10-7
## [49] abind_1.4-8 httr_1.4.8 compiler_4.5.1
## [52] withr_3.0.2 S7_0.2.1 BiocParallel_1.44.0
## [55] fastDummies_1.7.5 MASS_7.3-65 tools_4.5.1
## [58] lmtest_0.9-40 otel_0.2.0 httpuv_1.6.16
## [61] future.apply_1.20.2 goftest_1.2-3 glue_1.8.0
## [64] nlme_3.1-168 promises_1.5.0 grid_4.5.1
## [67] Rtsne_0.17 cluster_2.1.8.2 reshape2_1.4.5
## [70] generics_0.1.4 gtable_0.3.6 spatstat.data_3.1-9
## [73] tidyr_1.3.2 data.table_1.18.2.1 stringfish_0.18.0
## [76] XVector_0.50.0 spatstat.geom_3.7-0 BiocGenerics_0.56.0
## [79] RcppAnnoy_0.0.23 ggrepel_0.9.7 RANN_2.6.2
## [82] pillar_1.11.1 stringr_1.6.0 spam_2.11-3
## [85] RcppHNSW_0.6.0 later_1.4.7 splines_4.5.1
## [88] dplyr_1.2.0 lattice_0.22-9 deldir_2.0-4
## [91] survival_3.8-6 tidyselect_1.2.1 Biostrings_2.78.0
## [94] miniUI_0.1.2 pbapply_1.7-4 knitr_1.51
## [97] gridExtra_2.3 IRanges_2.44.0 Seqinfo_1.0.0
## [100] scattermore_1.2 stats4_4.5.1 xfun_0.57
## [103] matrixStats_1.5.0 stringi_1.8.7 UCSC.utils_1.6.1
## [106] lazyeval_0.2.2 yaml_2.3.12 evaluate_1.0.5
## [109] codetools_0.2-20 tibble_3.3.1 cli_3.6.5
## [112] RcppParallel_5.1.11-1 uwot_0.2.4 xtable_1.8-8
## [115] reticulate_1.45.0 systemfonts_1.3.1 jquerylib_0.1.4
## [118] dichromat_2.0-0.1 Rcpp_1.1.1 GenomeInfoDb_1.46.2
## [121] spatstat.random_3.4-4 globals_0.19.1 png_0.1-9
## [124] spatstat.univar_3.1-6 parallel_4.5.1 pkgdown_2.2.0
## [127] dotCall64_1.2 sparseMatrixStats_1.22.0 bitops_1.0-9
## [130] listenv_0.10.1 viridisLite_0.4.3 scales_1.4.0
## [133] ggridges_0.5.7 purrr_1.2.1 crayon_1.5.3
## [136] rlang_1.1.7 qs2_0.1.7 cowplot_1.2.0
## [139] fastmatch_1.1-8